










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO  uBio
uBio
Share

 Permalink
PermalinkRevista Boliviana de Química
On-line version ISSN 0250-5460
Rev. Bol. Quim vol.35 no.3 La Paz 2018
SHORT REVIEW
Obtaining of alkenes by reductive coupling of carbonylic compounds; synthesis of Z,E-6-dodecene, syntheses of flexibilene and isocaryophyllene, mechanistic views; the organic chemistry notebook, N ° 14
Obtención de alquenos por acoplamiento reductivo de compuestos carbonílicos; síntesis de Z,E-6-dodeceno, síntesis de flexibileno y isocaryophyllene, vistas mecanicistas; el cuaderno de químicaorgánica, N ° 14
José A. Bravo1,*, José L. Vila2
1 Natural Product Laboratory, Phytochemistry, Chemical Sciences Department, School of Pure and Natural Sciences FCPN, Universidad Mayor de San Andrés UMSA, P.O. Box 303, Calle Andrés Bello s/n, Ciudad Universitaria Cota Cota, phone +59122792238, La Paz, Bolivia, jabravo@umsa.bo, joseabravo@outlook.com, www.umsa.bo
2Natural Product Laboratory, Green Chemistry, Chemical Sciences Department, School of Puré and Natural Sciences FCPN, Universidad Mayor de San Andrés UMSA, P.O. Box 303, Calle Andrés Bello s/n, Ciudad Universitaria Cota Cota, phone +59122772269, La Paz, Bolivia, jlvila@umsa.bo, www.umsa.bo
*Corresponding author: jabravo@umsa.bo , joseabravo@outlook.com
Received 08 12 2018 Accepted 08 25 2018
Abstract
The Organic Chemistry Notebook Series, a Didactical Approach, is the series designed with educational purposes in the organic synthesis field. With the present paper we add to a total of fourteen contributions so far in the series.
This series of studies is designed to help students when getting started in the synthesis subject. The method of learning includes many fully and explicitly designed reactions step by step. The best manner to understand a synthesis is by means of graphical views which have been proposed by the authors of the series, and when they are accompanied in most of the cases by illustrative comments by the authors that describe de graphical mechanistic proposals and add some criteria deduced from the different mechanistic steps. We have taken a series of reactions compiled by W. Carruthers in 'Some modern methods of organic synthesis', and we have proposed didactical and mechanistic views for them. This theme is included in the chapter "Formation of carbon-carbon double bonds" in the mentioned text.
Carbonyl compounds can be reduced until alkenes by reductive dimerization, this is the so called Intermolecular Carbonyl Coupling InterMolCC. For aldehydes and ketones is feasible the obtaining of alkenes on reductive dimerization by using a reductive agent based on Ti(III) 3C1- and LiAlH4 or Zn(Cu) or active metallic Ti. The InterMolCC also includes the Intermolecular Mixed Carbonyl Coupling InterMolMCC. Also, we described in a mechanistic manner what has been exposed by diverse other authors with regard to a variant of InterMolMCC, the Intramolecular Mixed (or not) Carbonyl Coupling IntraMolMCC.
Keywords: Organic Chemistry, Alkenes, Reductive coupling, Carbonyl, Aldehyde, Ketone, Flexibilene, Z,E-6-dodecene, Isocaryophyllene, Mechanisms of Reactions, J.E. McMurry, W. Carruthers.
Resumen
"La serie de cuadernos de química orgánica, un enfoque didáctico", es la serie diseñada con fines educativos en el campo de la síntesis orgánica. Con el presente documento, sumamos un total de catorce contribuciones hasta ahora en la serie.
Esta serie de estudios está diseñada para ayudar a los estudiantes cuando se inician en el tema de síntesis. El método de aprendizaje incluye muchas reacciones diseñadas completa y explícitamente paso a paso. La mejor manera de entender una síntesis es por medio de vistas gráficas que han sido propuestas por los autores de la serie y cuando en la mayoría de los casos están acompañadas por comentarios ilustrativos de los autores que describen propuestas mecanicistas gráficas y agregan algunos criterios deducidos de los diferentes pasos mecanicistas. Hemos tomado una serie de reacciones compiladas por W. Carruthers en "Algunos métodos modernos de síntesis orgánica", y hemos propuesto puntos de vista didácticos y mecanicistas para ellos. Este tema se incluye en el capítulo "Formación de dobles enlaces carbono-carbono" en el texto mencionado.
Los compuestos carbonílicos se pueden reducir hasta alquenos por dimerización reductiva. Para los aldehidos y las cetonas es factible la obtención de alquenos en la dimerización reductora utilizando un agente reductor basado en Ti (III) 3C1- y LiAlH4 o Zn (Cu) o Ti metálico activado.
INTRODUCTION
Due to lack of knowledge of classical mechanisms, students experiment emptiness when comprehending a determined synthesis found in the literature. Since a mechanistic proposal is naturally mandatory for a rational explanation of producís emerging from a synthesis, we offer the present series theming on mechanistic approaches on several published syntheses. As academics we are committed with the didactics and we have designed a series of arricies exposing mechanistic theoretical proposals, arricies have a character of short review. The present contribution: Obtaining of alkenes by reductive dimerization of carbonylic compounds; mechanistic views; the organic chemistry notebook, n° 14, is the fourteenrh study in rhe series: "The Organic Chemistry Notebook Series, a Didactical Approach" [1-13].
REVIEW OF REACTIONS, MECHANISTIC THEORETICAL PROPOSALS, DISCUSSION
Reductive coupling of carbonyl compounds [14]
Aldehydes and ketones are appropriate substrates for transformation to afford alkenes [14]. The method includes reductive dimerization of substrates by means of carbonyl coupling; the employ of TÍCI3 and LiAlH4 or Zn(Cu) couple as reductive agents, is mandatory [14]. Alternatively, an active titanium metal species can be applied, the later can be formed by reducing TiCI3 with K or Li [14-17]. This reaction was widely used, however its utility is limired since when rhe inrermolecular reaction occurs, this lacks selectivity and affords mixtures of the E and Z isomers. See the example in in Figure 1.
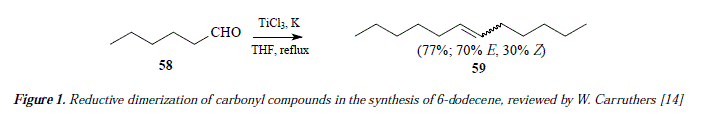
The reaction requires a surface of active titanium partióles for interactions [14]. Reaction occurs in two stages. In the first step a pinacol reduction happens to conduct the reaction to the apparition of a new C-C bond by mediating a monomeric coupling or dimerization [14]. Carbonyl is reduced into ketyl by means of receiving an electrón from the donor titanium; ketyl dimerizes to pinacol [14]. Pinacols have been proposed as intermediates since they convert into alkenes in smooth conditions when titanium is employed [14]. Subsequently, in the second step of the reaction, deoxygenation occurs, because of coordination of pinacol to titanium particle [14]. Excision of the two bonds O-C comes about, yielding the alkene along with the oxidized titanium surface [14]. See Figure 2 for an explanatory scheme of this reaction [14]. See figure 3 for a mechanistic view of reaction of Figure 1.
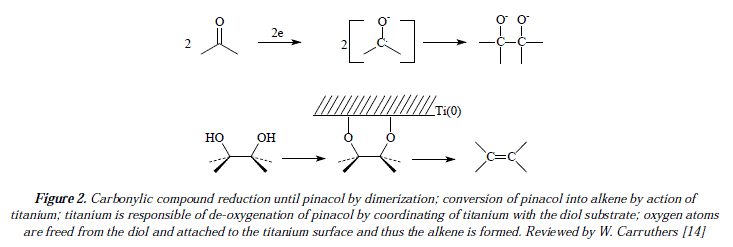

Comments
The dimerization and reduction of aldehydes into alkenes containing as double number of carbons in comparison to the aldehyde, can be achieved by the coupling of 2 molecules of aldehyde. In Figure 3 the two molecules of hexanal (58) are coplanar in the same plan one faced to the other in proximity enough to make possible coupling of the carbonyl groups but with oxygen of carbonyls placed as far as possible one from another. The pentyl moiety of the two hexanal molecules are very close each other because of the similarity of their polarity (these moieties are non-polar and they mix easily), this alkylic characteristic of hexanal makes possible the monomeric coupling. The current method under analysis is the one employing reduced titanium from TiCl3, and excludes the utilization of any reducing agent, namely LiAlH4 [14]. It is meant by that that we are in a surface catalysis process, which implies a radicáis' mechanism, whereas hydride using implies a SN2 ionic mechanism [14-17]. Hence, in Figure 3, the % electrons of both carbonyl groups of the aldehydes delocalize onto the extremes of the ancient double bond forming a di-radical. This di-radical is stabilized by pairing the unpaired oxygen electron with an electron on the Ti(0) which becomes thus a "prét a porter" electron. The departure of such electron from the surface of Ti(0) occurs by oxidizing to Ti(I) and increasing thus the electron richness of the electron-receiver oxygen. The whole process runs under the auspicious covalent bond establishment between oxygen from aldehyde and the titanium atoms, represented by segmented traces in Figure 3. Once the oxygen received the metallic electron, the former becomes an anión alkoxy, which converts to alcohol by interaction with water equilibrium, taking the aqueous proton. Titanium is now a Ti+ species, always imbibed in the Ti(0) surface. According to Figure 3, carbonyl has been reduced so far and sequentially to ketyl, alkoxy and presently to alcohol. Next step contemplates the reduction of alcohol to alkene by means of extracting oxygen from alcohol by eliminating gas hydrogen first followed by homolysis of the C-O, leading to an unsaturated hydrocarbon (E and Z alkene 59) and oxidized Ti+ coordinated to neutral oxygen atoms. Highly unstable due to its atom condition, or an only atom of oxygen with a double radical character (diradical), and because its high electronegativity, oxygen provokes an oxidation of Ti+ to Ti2+.
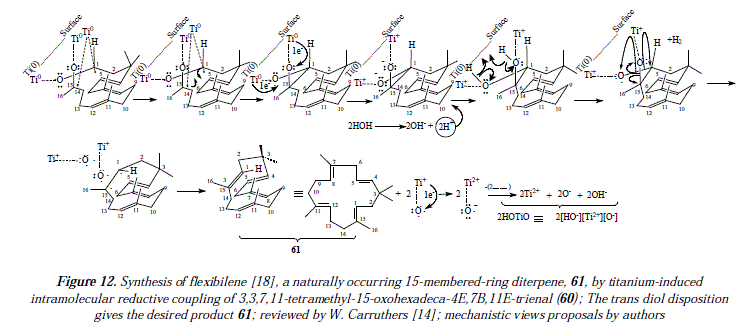
The preparation of alkenes of increased number of carbons is also feasible by combination of two different carbonyl compounds, ketone and aldehyde for instance [14]; mixtures of compounds are produced reducing its effectiveness and interest in synthesis [14]. One example of alkenes from condensation of two different carbonyl groups is an intramolecular reaction of a di-carbonyl compound to form a cyclic alkene [14]. Thus, alkenes of 4 to 16 atoms of carbón have been prepared by this method [14]. In this route, compound 60 (a keto-aldehyde) was employed to produce compound 61 named flexibilene [18], a cycled polyene (52%) [14]. See Figure 4 for the scheme of the reaction [14], and Figures 11 and 12 for a mechanistic explanation.
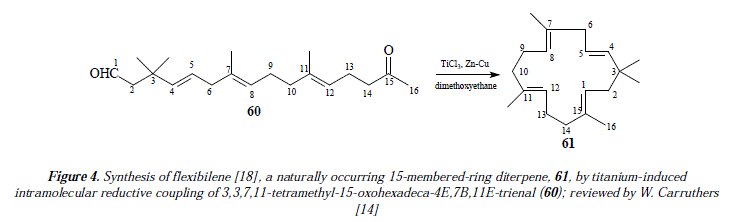
Comments
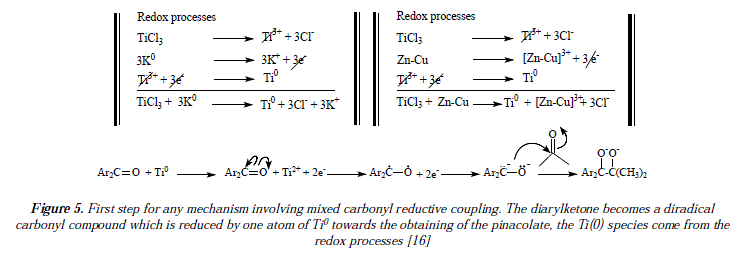
The mechanistic approaches for reductive couplings of carbonyls are scarce. The fundamental differences between approaches lie in the nature of the process, ionic, by radicals or mixed. According to the literature, it has been established that carbonyl mixed couplings are effected by means of the conversion of carbonyl into alcohol to afford a pinacol after condensation [19,20]. It has also been proposed and it is widely accepted that pinacol dimerization occurs vía anion radicals [21]. It has been proposed that the mixed carbonyl coupling of diaryl ketones with acetone happens in such a way that diaryl ketone reduces to a dianion whose carbanion realizes a nucleophilic attack over carbonyl of acetone to afford the pinacolate dianion [16]. Figure 5 shows this feature [16], this is the beginning of any further mechanistic proposal [16]. Figures 6, 7 and 9 show four of many possible mechanistic routes for mixed carbonyl reductive coupling [16]. Figures 6, 7 and 9 are schemes for dimerization of carbonyl compounds to afford olefins which present paths Al, A2, B and C as possible mechanisms [16]. The initial or first step (Fig. 5) is the carbonyl coupling to form the pinacolate after reduction by Ti(0) [16]. In Path Al pinacolate forms a five membered ring between oxygen atoms and the same titanium atom. Homolytic C-0 bonds excision follows to form a hydrocarbon the alkene C=C bond and the diradical [TiO2] Titanium (II) further oxidizes into Ti(IV) by providing two electrons to form the % bonds Ti(IV)(=O)2. The principal feature of path A1 is the fact that it is a concerted process where homolysis, metal oxidation and new bonds formation are simultaneous. The proposition of an only titanium atom bonding to two oxygen atoms makes the reaction regioselective. Figure 3 which is a dimerization of hexanal, clearly shows in the producís that the reaction is not regioselective which means that Al is not a likely mechanism in this case. However, a hypothesis for the observed mixture of cis and trans producís backed by reports on pinacol reverse reaction could justify the stereoisomers mixture found as producís [22,23]. The non-concerted mechanistic alternative is path A2 (Figure 6, [16]).
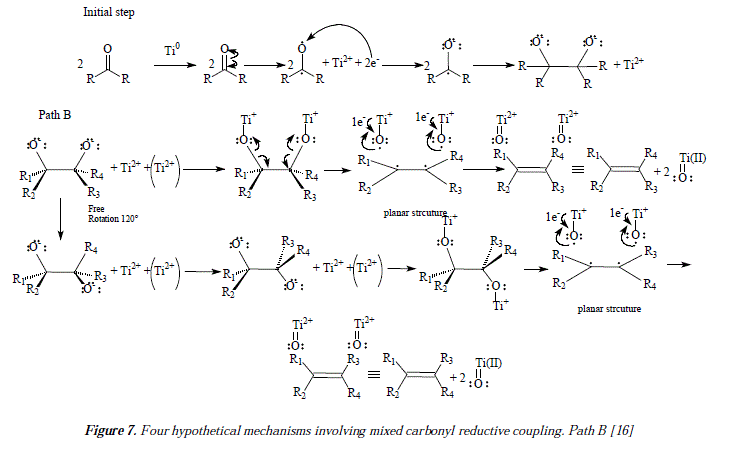
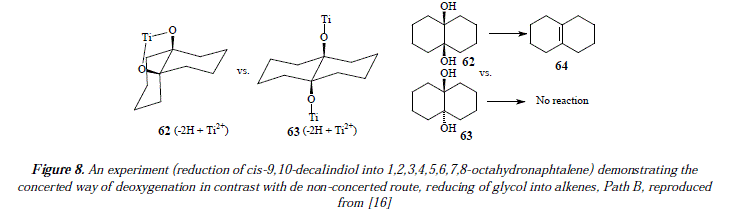
Such a fact has been proved experimentally by the reducing of meso- and dl-5,6-dihydroxydecane with TiCl3/K to obtain Z- and E-5-decene [16]. Another mechanistic possibility is depicted in path B of Figure 7 [16]. Path B shows the absence of the five-membered ring, and the presence of two titanium atoms instead of one as shown in paths A1 and A2, involved in the reductive process [16]. The process is supposed to not to be concerted [16]. An experiment has been designed to prove that path B is not the more appropriate mechanism, at least in the case where cis- and trans-9,10-decalindiol (62 and 63) was reduced to 1,2,3,4,5,6,7,8-octahyronaphtalene (64) [16]. In such experiment the cis isomer afforded the naphthalene derivative suggesting a concerted reaction pathway but the trans isomer gave no alkene [16]. These empiric conclusions seem to point a concerted reaction pathway as Al instead of a non-concerted one or B [16]. See Figure 8.
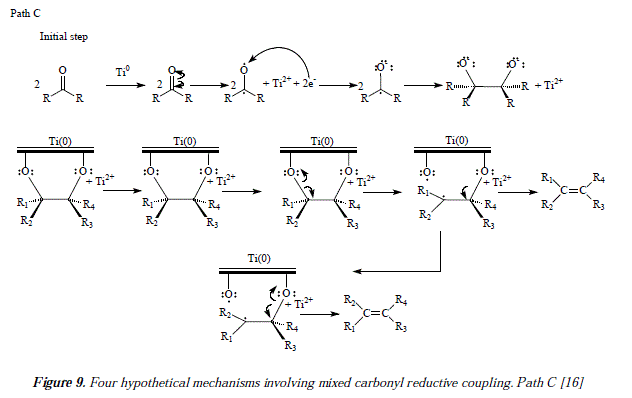
The last considered likely mechanism in the reference 16 is pathway C [16] (Figure 9). This implies the use of Ti(0) but in a heterogeneous catalysis process. The difference between paths C and A is that the two oxygens are linked to a Ti surface (path C) instead of a common Ti atom (path A) [16]. Both mechanisms are compatible with the results of the isomeric decalindiol reactions [16]. There has been designed an experiment mentioned by McMurry et al. [16] to establish the presence or the absence of a five-membered ring intermedíate that showed a much faster reaction of cis-diol 65 than trans-diols 66 and 67 with various reagents assumed to form five-membered rings like lead tetraacetate for instance. The kinetic studies carried out demonstrated the easy (rapid) way for glycol 65 to form a five-membered lead alkoxide and the hard, even impossible way (too slow to be kinetically monitored) to form it for 66 and 67 [16]. The reason is simple, on the one hand, the close to position, of hydroxyls of 65 with regard to the metal surface, and on the other hand, the far away from position, of hydroxyls toward the metallic surface in 66 and 67. Thus, a five-membered ring is not feasible for 66 and 67 under the interaction with lead tetraacetate [16]. Let us return to the interaction with titanium now. Similar kinetic studies, employing titanium in place of lead tetraacetate, have been done by McMurry et al. [16]. The results are conclusive. The employ of cis- (65) and trans-camphanediols (66 and 67) conducted to the reduced 2-bornene [16] (Figure 10).
Literally we reproduced a fragment as an excerpt that better explains the conclusions of McMurry and collaborators about the choice of path C in place of path A for proposing a mechanism for the reactions under current survey: "If the titanium deoxygenation reaction also proceeds through a five-membered ring (path A), we would expect cis diol 7 (65) to reduce at a much faster rate than trans diols 8 (66) and 9 (67). If, however, the hydroxyls need only approach a common broad surface (path C), both cis and trans diols should reduce. Treatment of cis diol 7 (65) and a 70:30 mixture of trans diols 8 (66) and 9 (67) in side by side experiments gave the results shown in Table IX. Aliquots were periodically removed, and yields of 2-bornene were determined by GLC. The table clearly shows that both cis- and trans-camphanediols were reduced at approximately the same rate and were complete after 5 h. These results strongly suggest to us that the deoxygenation of diols does not require the formation of a five-membered ring intermedíate. We therefore conclude that the reaction occurs by the route shown in path C of Scheme II; i.e., the deoxygenation of diols occurs in a heterogeneous process on the surface of an active titanium particle (see Scheme III)" (reproduced from [16]).
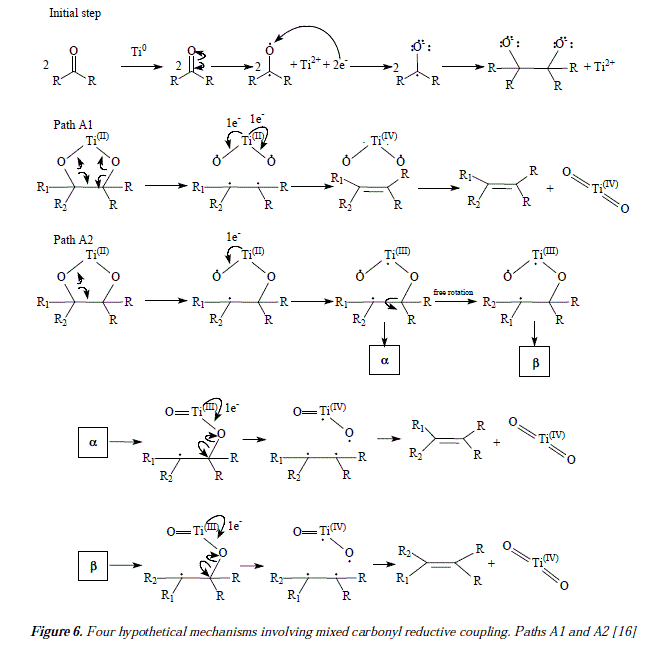
Our own approach to the problematic includes mechanistic proposals as the ones showed in Figure 3 and Figures 11 and 12, that correspond to the synthesis of flexibilene [14] (Figs. 12 and 13) and oí Z- and .E-6-dodecene [14] (Figure 3), which were not studied by McMurry and col. in reference 16. In this arricie we tried to give complementary ideas on the path C, now collectively accepted as the most appropriate mechanism for carbonyl couplings, with some, so far missing concepts, regarding the process itself. However, our focus is strictly theoretical, but it adds in some cases, and replaces in others, to what was already established my McMurry and col. [16]. Our mechanisms (Fig. 3, 11 and 12) propose, instead of the radical-anionic initial step as exposed in Figures 5-7 and 9 and in reference 16, a purely radical initial process. Figure 9 shows some missing explanation with respect to the cation Ti2+, which seems not to be equaled in a strict stoichiometrical sense. Another feature of mechanisms in Fig. 3, 11 and 12, is the fact of considering two or more atoms of Ti(0) involved, a fact not specifíed in path C by McMurry and col. In this way, we propose that the carbonyls coupling just beíore reduction into olefins occurs on tñe metal suríace ítselí and not before intervention of the surface as showed by McMurry and col. in Figure 5 [16]. A missing explanation in path C (Figure 9) is about the number of titanium atoms in the surface involved in coupling with hydroxyls in the carbonyls' reduction [16]. We propose two superficial atoms of Ti(0) that oxidize later when going toward producís. Also a missing explanation in [16] is the manner how titanium atoms interact with oxygen of pinacol; is it a covalent bonding or a coordination bonding? Questions that remain unanswered. Thus, we propose covalent bonding.
In Figure 3 we propose essentially that the metallic surface keeps reduced except for a couple of titanium atoms that oxidize by giving one electrón each to an oxygen in the substrate to afterward quit the surface at any moment under the form of Ti(I). But we believe that these oxidized titanium atoms, belong still for a while to the conglomérate of the surface, in a continuous interaction with the substrate in the formation of the σ bond first, and until deoxygenation to form the π bond of the final olefinic product. Once these cations of Ti+, bonded to an oxygen, have abandoned the metallic platform, they oxidize to Ti2+ by giving one electron to oxygen which becomes an anion (O-). A third titanium atom is involved by temporarily coupling the two radicals of the intermedíate with available electrons in the titanium atom. Such interaction lasts in the way of a surface catalysis, until the formation of the sigma bond between the oxymethine groups in the synthesis of compound 59, and between the methylcarboxy and the oxymethine groups in the synthesis of 61. In the synthesis of flexibilene (61, Figures 11 and 12), we apply the same succession of steps as the one described for the synthesis of 6-dodecene.
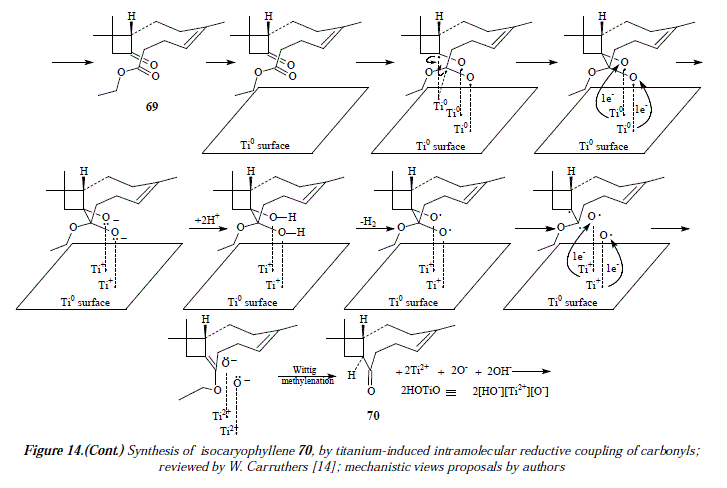
Figure 11 is a first approach to the proposal of a mechanism for the synthesis of compound 61. As already mentioned, we apply the same steps developed for the mechanism of 59. This supposes the interaction between the substrate the ketoaldehyde 60 and a surface of Ti(0). Thus, the surface interacts with the substrate throughout three titanium atoms (Fig. 11 A), whose highly directional atomic orbital (hybrid or not) should overlap with a sp3 orbital of radical oxygen (or a sp2 of radical carbon) containing a single electron. This interaction constitutes a covalence or a covalent Ti-O (or Ti-C) bonding containing a couple of electrons one from titanium and the other from radical oxygen (or the radical carbon). Let us signal at this point that the interaction Ti-C requires two highly directional atomic orbitals of titanium, each containing an unpaired electron (Fig. 11 A B). Following steps in Figure 11, when titanium (0) transfers its covalent bonding electron to oxygen (Fig. 11 C), the covalence becomes an ionic bond.

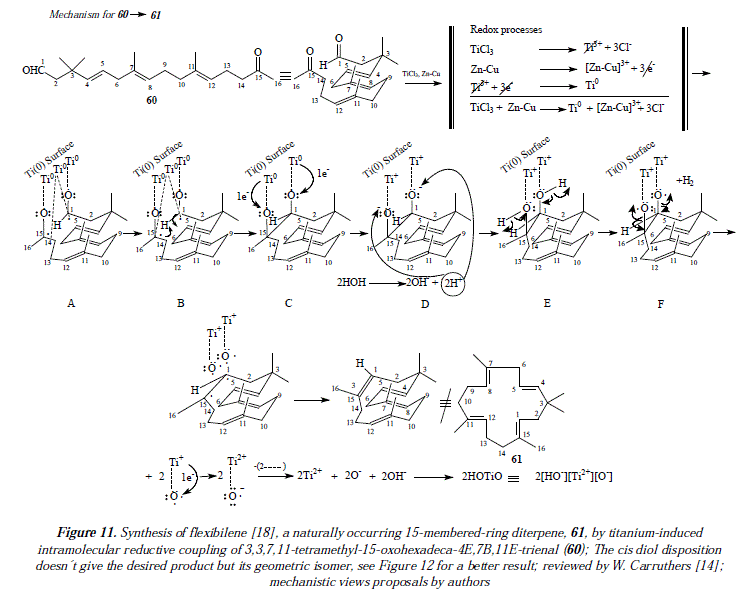
Titanium atoms are now cations and oxygen atoms are anions and they're still linked as an ionic pair or an ionic bond (Fig. 11 D). In Figure 11 E when the alkoxides RO- become a secondary and a tertiary alcohol by protonation from water, the interaction of titanium in the surface (as Ti+) and oxygen in the substrate passes by the overlapping of the very same titanium atomic orbital originally used in the covalences with carbonyl's oxygens, but empty of electrons and a sp3 orbital with two electrons available of oxygen who shares partially the cationic charge of titanium+ which reduces to Ti(0) in equilibrium (HO-Ti+↔HO+-Ti) with the equilibrium displaced toward the cationic Ti+ ion (HO-Ti+). This is the driving force that provokes deoxygenation of the substrate by homolysis of C-0 bonds. The interaction surface-substrate ends in Fig. 11 F when deoxygenation occurs and the alkene is formed.
The inconvenient in Figure 11 is that that disposition of titanium atoms (cis-diol), side by side in the metallic surface, conducís to the cis H-I/CH3-16 isomer which is not the diterpene flexibilene (61), but its geometric isomer, whereas the trans H-I/CH3-16 isomer explained mechanistically in Figure 12 fits well the structure of flexibilene (61) and it's obtained from titanium atoms in mutual remote position (transs-diol). This last statement is supported by what is explained in Figure 10 regarding the surface catalysis, a mechanism currently accepted as the more adequate one in carbonyls' coupling to form olefins [16].
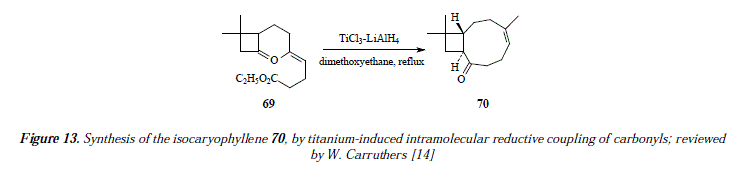
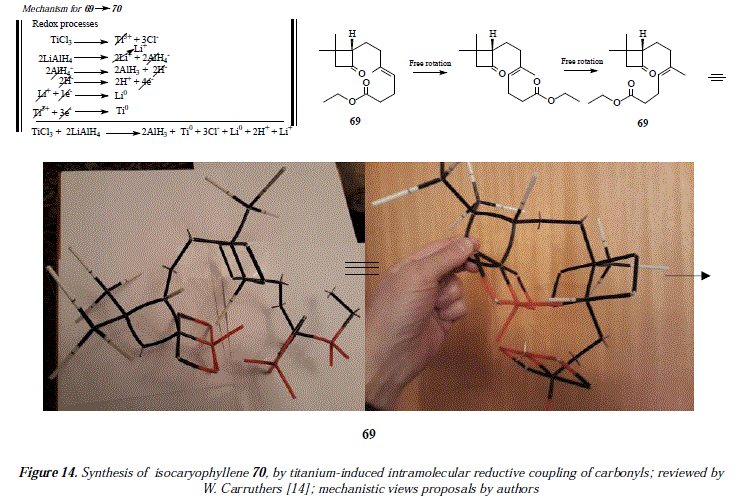
To end this contribution, we'll discuss the formation of cyclic ketones starting from keto-esters [14]. For example, isocaryophyllene (70) is synthesized from the keto-ester (69) followed by Wittig methylenation [14], Figure 13. The corresponding mechanism is exposed in Figure 14.
ACKNOWLEDGEMENTS
The authors express their gratitude to Prof. Eduardo Palenque, for bibliographic support, Department of Physics, and Prof. J. Mauricio Peñarrieta, for fruitful comments, Department of Chemical Sciences, Universidad Mayor de San Andrés.
REFERENCES
1. Bravo, J. 2005, The organic chemistry notebook series, a didactical approach. Theoretical mechanistic approach to diasteroselective synthesis of cis-1,2-dialkenylcyclopropanols and subsequent oxy-Cope rearrangement by Jin Kun Cha et al, Rev.Bol Quim.,23(1), 1-10. [ Links ]
2. Bravo, J.A., Mollinedo, P., Peñarrieta, J.M., Vila, J.L. 2013, Mechanistic views of intramolecular hydroxycyclopropanation of »-vinyl carboxylic esters, Rev. Bol. Quim., 30(1), 24-41. [ Links ]
3. Bravo, J.A., Vila, J.L. 2014, Mechanistic views of stereoselective synthesis of tri and tetra-substituted alkenes, part I; the organic chemistry notebook series, a didactical approach, n° 3. Rev. Bol. Quim., 31 (1), 61-67. [ Links ]
4. Bravo, J.A., Vila, J.L. 2015, Mechanistic views of stereoselective synthesis of tri and tetra-substituted alkenes, part II; the organic chemistry notebook series, a didactical approach, n° 4, Rev. Bol. Quim., 32(1), 15-23. [ Links ]
5. Vila, J.L., Bravo, J.A. 2015, Synthesis of alkenes by fragmentation reactions; Mechanistic views; the organic chemistry notebook series, a didactical approach, n° 5, Rev. Bol. Quim., 32(2), 37-44. [ Links ]
6. Bravo, J.A., Vila, J.L. 2015, Synthesis of alkenes by oxidative decarboxylation of carboxylic acids; Mechanistic views; the organic chemistry notebook series, a didactical approach, n° 6, Rev. Bol. Quim., 32(3), 45-52. [ Links ]
7. Bravo, J.A, Vila, J.L. 2015, Synthesis of alkenes from ketones via arylsulphonyl-hydrazones; mechanistic views; the organic chemistry notebook series, a didactical approach, n° 7, Rev. Bol. Quim., 32(4), 82-89. [ Links ]
8. Bravo, J.A., Vila, J.L. 2015, Stereospecific synthesis of alkenes from 1,2-diols; mechanistic views; the organic chemistry notebook series, a didactical approach, n° 8, Rev. Bol. Quim., 32(5), 121-125. [ Links ]
9. Bravo, J.A., Vila, J.L. 2016, Synthesis of alkenes by Claisen rearrangement of allyl vinyl ethers, part I; mechanistic views; the organic chemistry notebook series, a didactical approach, n° 9, Rev. Bol. Quim., 33 (1), 27-33. [ Links ]
10. Bravo, J.A, Vila, J.L. 2016, Synthesis of alkenes by Claisen rearrangement of allyl vinyl ethers, part II; mechanistic views; the organic chemistry notebook series, a didactical approach, n° 10, Rev. Bol. Quim., 33 (2), 95-103. [ Links ]
11. Bravo, J.A., Vila, J.L. 2016, Synthesis of alkenes by Claisen rearrangement of allyl vinyl ethers, part III; mechanistic views; the organic chemistry notebook series, a didactical approach, n° 11, Rev. Bol. Quim., 33(3), 127-133. [ Links ]
12. Bravo, J.A., Vila, J.L. 2017, Claisen rearrangement of allyl vinyl ethers to afford alkenes, part IV; mechanistic theoretical proposals; the organic chemistry notebook series, a didactical approach, n° 12, Rev. Bol. Quim., 34 (2), 40-49. [ Links ]
13. Bravo, J.A., Vila, J.L. 2017, Mechanistic theoretical proposals for: alkenes by claisen rearrangement of alfa-allylthio carbenes; aza-cope rearrangement of 4-butenyliminium ions; 2-substituted pyrrolidine derivatives; synthesis of perhydrogephyrotoxin, key step; part v; the organic chemistry notebook, n° 13, Rev. Bol. Quim., 34 (5), 142-149. [ Links ]
14. Carruthers, W. Some Modern Methods of Organic Synthesis, Cambridge University Press, 3rd ed., 1987, Worcester, U.K., pp. 180-182. [ Links ]
15. McMurry, J.E. 1983, Titanium-induced dicarbonyl-coupling reactions, Acc. Chem. Res., 16(11), 405-411. [ Links ]
16. McMurry, J.E., Fleming, M.P., Kees, K.L., Krepski, L.R. 1978, Titanium-induced reductive coupling of carbonyls to olefins, J. Org Chem., 43(11), 3255-3266. [ Links ]
17. Dams, R., Malinowski, M., Westdorp, I., Geise, H. 1982, On the mechanism of the titanium-induced reductive coupling of ketonesto olefins, /. Org. Chem., 47(2), 248-259. [ Links ]
18. McMurry, J.E., Matz, J.R., Kees, K.L., Bock, P.A. 1982, Synthesis of flexibilene, a naturally occurring 15-membered-ring diterpene, Tetrahedron Letters, 43 (17), 1777-1780. [ Links ]
19. McMurry, J.E., Flemming, M.P. 1974, New method for the reductive coupling of carbonyls to olefins. Synthesis of beta-carotene, J. Am. Chem. Soc, 96(14), 4708-4709. [ Links ]
20. McMurry, J.E., Flemming, M.P. 1976, Improved procedures for the reductive coupling of carbonyls to olefins and for the reduction of diols to olefins, J. Org. Chem., 41 (5), 896-897. [ Links ]
21. House, H.O., Modern Synthetic Reactions, W.A Benjamín, 2nd ed, 1972, New York, U.S.A., pp 167-169. [ Links ]
22. Schlosser, M., Weiss, P. 1970, Synthesis, 257. [ Links ]
23. Sharpless, K.B., Flood, T.C. 1972, J. Chem. Chem. Commun. 372. [ Links ]

