










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
 Citado por SciELO
Citado por SciELO Links relacionados
 Similares en
SciELO
Similares en
SciELO  uBio
uBio Compartir

 Permalink
PermalinkRevista Boliviana de Química
versión On-line ISSN 0250-5460
Rev. Bol. Quim vol.32 no.3 La Paz set. 2015
ARTÍCULOS ORIGINALES
Synthesis of Alkenes by oxidative decarboxylation of carboxylic acids; mechanistic views; the organic chemistry notebook series, a didactical approach, N° 6
José A. Bravo, José L. Vila*
Department of Chemistry, Laboratorio de Síntesis y Hemisíntesis, Laboratorio de Fitoquímica, Instituto de Investigaciones en Productos Naturales IIPN, Universidad Mayor de San Andrés UMSA, P.O. Box 303, Calle Andrés Bello s/n, Ciudad Universitaria Cota Cota, Tel. 59122792238, La Paz, Bolivia. joselu62@hotmail.com, jabravo@umsa.bo
*Corresponding author: joselu62@hotmail.com
Abstract
This is the sixth chapter in the series published by the same authors: "The Organic Chemistry Notebook Series, a Didactical Approach".
Here we offer the mechanistic views of the synthesis of alkenes by oxidative decarboxylation of carboxylic acids. The aim of this series of studies is to help students to have a graphical view of organic synthesis reactions of diverse nature. The oxidative decarboxylation of carboxylic acids is a useful method for generating alkenes. Here we propose the mechanism and its discussion for the application of the method of decarboxylation of diacids lacking nearby double bonds. Also, the route is explained mechanistically for the preparation of Dewar benzene. The thermal or photolytic decomposition of di-t-butyl per-esters is described. The treatment of monocarboxylic acids to afford alkenes in the presence of lead tetraacetate and copperII acetate is briefly discussed. The alkylation-decarboxylation of aromatic acids is also explained. The oxidative decarboxylation of carboxylic acids can eventually conduct to the obtaining of ketones instead of alkenes. We have used a series of reactions reviewed by W. Carruthers, and we have proposed didactical and mechanistic views for them. This latest approach is included in the synthetic methods reviewed by W. Carruthers with respect to the "Formation of carbon-carbon double bonds". Spanish title: Síntesis de alquenos por descarboxilación de ácidos carboxílicos; vistas mecanísticas; De la serie: El cuaderno de notas de química orgánica, un enfoque didáctico, N°6.
Keywords: Organic Chemistry, oxidative decarboxylation, carboxylic acids, Alkenes, Mechanisms of Reactions.
MECHANISTIC PROPOSALS AND DISCUSSION
Decarboxylation ofvicinal diacids lacking nearby double bonds
As academics we are concerned with the didactical importance of covering the needs of debutant students in organic synthesis. This is the sixth study in: "The Organic Chemistry Notebook Series, a Didactical Approach" [1-5]. The method of decarboxylating carboxylic acids has proven to be useful [6]. Variations on the basic steps have been introduced essentially for the improvement of the yields [6]. The aim is the generation of double C-C bonds starting with vicinal dicarboxylic acids and subsequent decarboxylation, as schematically viewed in Fig. 1 [6,7].
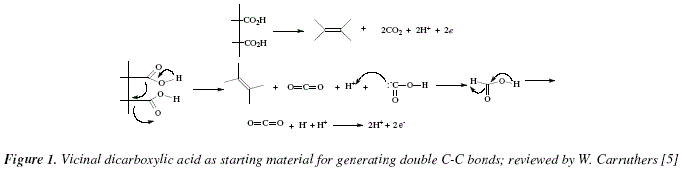
The treatment with lead tetraacetate in boiling benzene can be improved with the presence of oxygen becoming thus applicable for vicinal dicarboxylic acids lacking nearby double bonds [6] (Fig. 2).

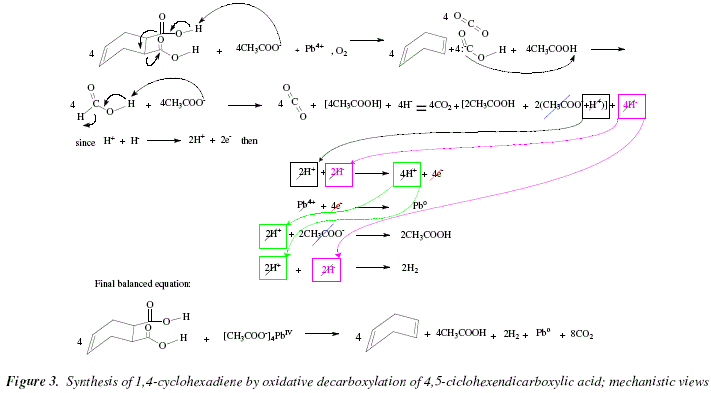
The mechanism is seen in Fig. 3. In step 1, lead acetate pulls off a proton from one of the carboxylic acid residues of the substrate to form a carboxylate residue. What follows is the immediate excision of the bonding of the substituent carboxylate. This electronic movement evokes somehow the Diels Alder mechanism with respect to the formation of the new double bond. The contrast is the rupture of both sigma bonds instead of the transformation of two double into single bonds in Diels Alder. However, indeed, this is not a pericyclic reaction. Besides cyclohexenone, CO2 and carbanionic carboxylic acid evolve from this fragmentation. The acid-base interaction between the acetic acid formed from the lead tetraacetate and the carbanionic carboxylic acid gives formic acid and tetraacetate is regenerated. In step 2, tetraacetate pulls off a proton from formic acid provoking decarboxylation (generation of CO2) and acetic acid plus 4 hydrides. In step 2, two of the four acetic acid equivalents ionize and generate two acetate and two proton equivalents. These two proton equivalents plus two from the four hydride equivalents of step 2 generate 4 protons and 4e- due to oxidation of hydride in step 3. In step 4, PbIV (Pb4+) is reduced to Pb0 thanks to the previous oxidation step that provided four e-. In step 5, two of the four proton equiv generated in step 3 interact with two acetate residues to generate 2 acetic acid equiv. Step 6 is the covalence formation between 2H+ and 2H-generating H2.
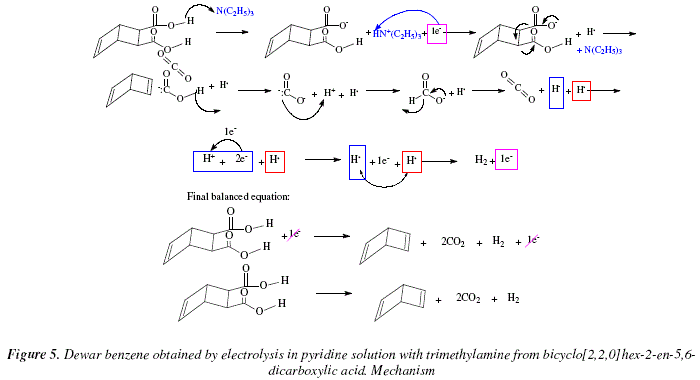
An interesting procedure with good yields is the obtaining of dienes by electrolysis of the dicarboxylic acid in pyridine solution with trimethylamine added. The reaction goes on in mild conditions, and the method is appropriate "for preparing highly strained unsaturated small and bridged ring compounds" [6]. As an example let's mention the preparation of Dewar benzene frombicyclo[2,2,0]hex-2-en-5,6-dicarboxylic acid, Fig. 4 [6].

In the first step an acid reaction occurs with the liberation of a proton from one of the acid residues of bicyclo[2,2,0]hex-2-en-5,6-dicarboxylic acid. Triethylamine serves as temporary base to accommodate this proton but soon after the proton is partially reduce by 1e- to H. Also, decarboxylation occurs with the first carboxylate residue to form the alkene and expel the second carboxylic acid residue under the form of carbanion. In the second step, the carbanionic carboxylic acid frees its proton which neutralizes the carbanion to afford formiate. Formiate expels a hydride and generates an equivalent of carbon dioxide. In the third step the hydride oxidizes to give a proton and two electrons, one of which generates a second .H species. The .H species overlap each other and form gaseous hydrogen. This step regenerates the electron employed in the electrolytic method under review. See Fig. 5 for mechanistic proposal.
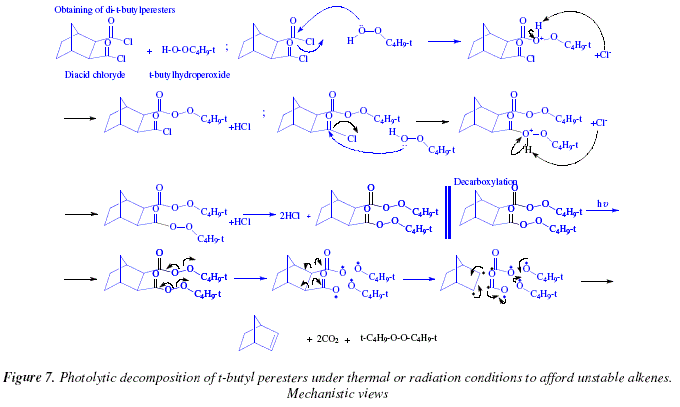
An easy and useful method, done under mild conditions, and activated under thermal or radiation conditions to provoke photolytic decomposition, uses t-butyl peresters, obtained at its turn from diacid chlorides and t-butyl hydroperoxide, Fig. 6 [6].
This photolysis reaction is applicable for synthesizing thermally unstable alkenes [6]. The vicinal dicarboxylic acids employed in this reaction can be obtained by Diels Alder reaction [6]. An alternative is the photosensitised addition of maleic anhydride to dienes or alkenes [6].
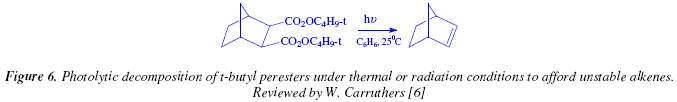
The obtaining of the t-butyl peresters is effected by nucleophilic substitution using diacid chloride as starting substrate and t-butylhydroperoxide as nucleophile. The outer oxygen of one equivalent of the two t-butylhydroperoxide equivalents acts as nucleophile over one of the acid chloride function of the bicyclo-compound. This forms a mono-t-butylperester derivative protonated. The second step is the substitution on the second acid chloride extreme of the bicyclo-compound to afford a di-t-butylperester. What follows is the irradiation of the di-t-butylperester which provokes homolytic rupture of the weakest bonding in the substrate, namely the peroxide bridge. This fact generates two single radicals (t-butoxy) and a double radical (di-carboxylate radical). This extremely reactive species (the double radical), collapses into another double radical (proto-alkene) and two carbonic anhydride residues. The two single radicals (t-butoxy) prefer to couple each other to generate a t-butoxyperoxide molecule. See Fig. 7 for this mechanism.
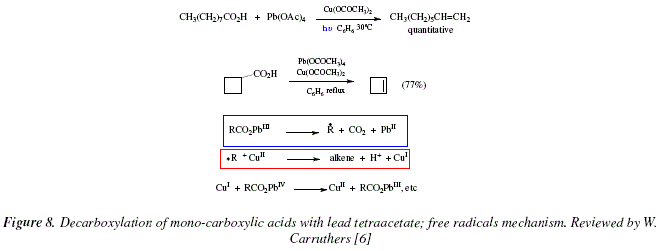
The oxidative decarboxylation of mono-carboxylic acids is a related reaction to the previously described reactions, and this is effected under treatment with lead-tetraacetate [6]. The development of the reaction depends on the structure of the acid giving poor yields and mixture of products, this, under normal conditions [6]. However, with the addition of catalytic amounts of copperII acetate the increasing of the yields of alkenes synthesized is significant [6]. The reaction is applied to primary and secondary carboxylic acids, either under photolysis, or under thermolysis, in benzene at its boiling point [6]. For example, nonanoic acid can be transformed into 1-octene in high yields and cyclobutanecarboxylic acid affords cyclobutene in 77 % yield. A mechanism by free radicals has been proposed by authors [6]. See Fig. 8.
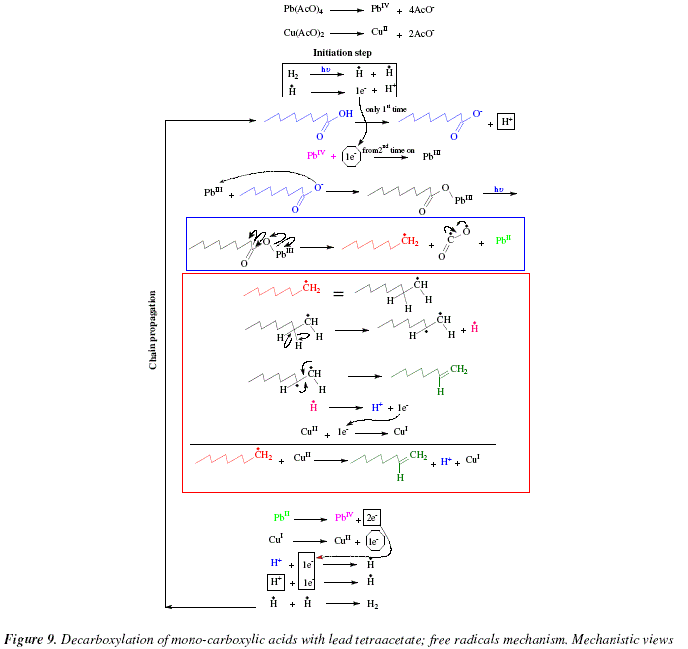
The mechanism is shown in Fig. 9. The acetates of lead and copper dissociate to give PbIV and CuII. Under the limited information offered by the review, we proposed a mechanism including radical formation. The mechanism is composed of two steps: initiation step and chain propagation. The initiation step (that is not mentioned in the review [6]) is assumed here as addition of a catalytic amount of H2 to the reacting mixture and subsequent photolysis giving rise to the radical H. and its oxidation to afford an initial electron to be employed in the chain-propagation during the first reduction of PbIV to PbIII. During the loop, this electron is provided by oxidation of CuI to CuII. The chain propagation includes a first step of carboxylic acid dissociation to acetate and H+ followed by reduction of PbIV to PbIII with the electron provided soon after by the oxidation of CuI to CuII. The acetate and the PbIII they form an adduct; this adduct absorbs radiation (hv) and gives rise to R., CO2 and PbII. The alklyl R. becomes an alkene and an H radical. This H radical is oxidized to H+ to provide the electron to reduce CuII into CuI. The complementary part of the chain propagation is the oxidation of PbII into PbIV and CuI into CuII and the generation of H2. After this, the cycle reinitiates at the stage of the dissociation of the carboxylic acid. This mechanistic proposal is highly limited and restrained by certain lack of information which is unavailable in the review [6], and could be improved after proper consultations with the correct bibliographic source. Let us remark with respect to Fig. 8 and 9, that those reactions inside the blue and red square in Fig. 8, are mechanistically explained in squares of the corresponding color in Fig. 9.
As an example of the applications of this reaction let's cite the alkylation-decarboxylation of aromatic acids. Fig. 10.
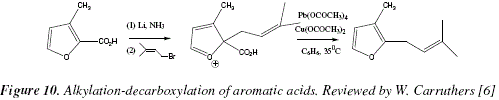
The lithium and ammonia employed provoke somehow delocalization of one of the aromatic ![]() systems until locating the negative charge over the carbonyl oxygen of the aromatic acid. The direct alkylation occurs when the carbanion attacks the alkyl bromide in a SN2 manner [6]. The furan derivative, rosefuran can be obtained following the oxidative radical process of Fig. 10 and 11 [6].
systems until locating the negative charge over the carbonyl oxygen of the aromatic acid. The direct alkylation occurs when the carbanion attacks the alkyl bromide in a SN2 manner [6]. The furan derivative, rosefuran can be obtained following the oxidative radical process of Fig. 10 and 11 [6].
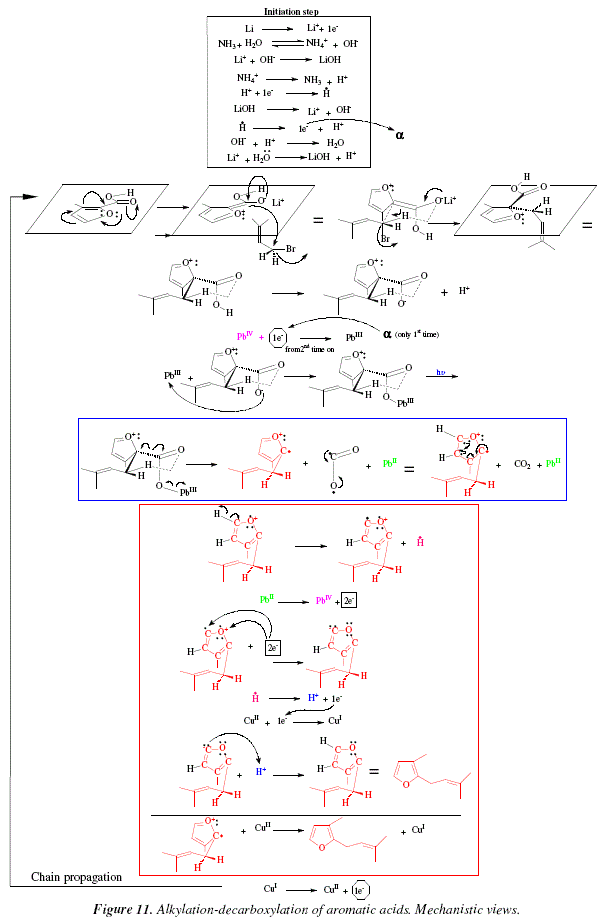
These two last mechanisms (Fig. 9 and 11) although presenting some species stoichiometrically unjustified (H+ and AcO-), give at least a global idea of the movement of species as well as the oxidation-reducing processes.
A carboxylic acid can be converted into a ketone instead of an alkene following oxidative decarboxylation. Carbon loss is verified during the process [6]. The acid converted in a dianion suffers sulphenylation under dimethyl disulphide [6]. The sulphenylated acid reacts with N-chlorosuccinimide and gives a diacetal with loss of carbon anhydride adding sodium bicarbonate [6]. The diacetal goes a hydrolysis to afford the ketone at 0°C [6]. The acid can be synthesized from cyclohexadiene and acrylic acid; this last is equivalent to the ketene in this homologue of the Diels Alder reaction [6]. See Fig. 12.
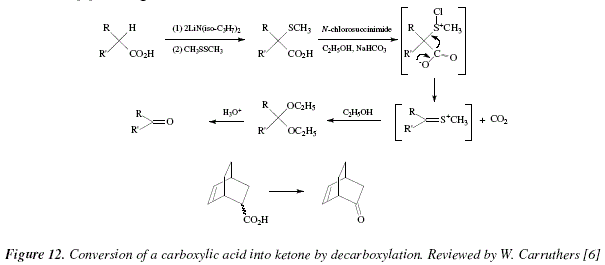
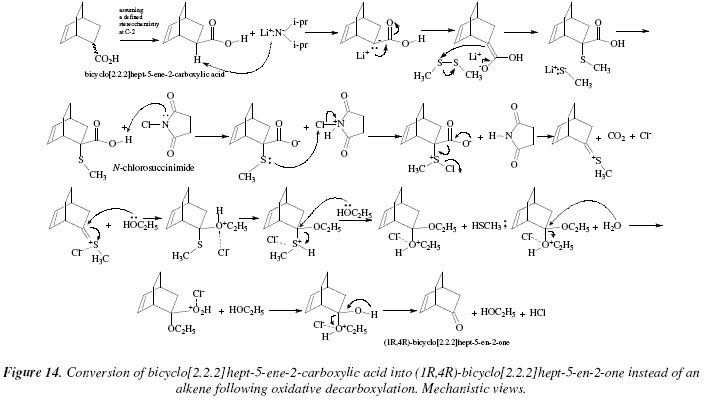
Another example is shown in Fig. 12 with the decarboxylation of bicyclo[2.2.2]hept-5-ene-2-carboxylic acid into (1R,4R)-bicyclo[2.2.2]hept-5-en-2-one [6]. The mechanistic views corresponding to Fig. 12 are exposed in Fig. 13 and 14.
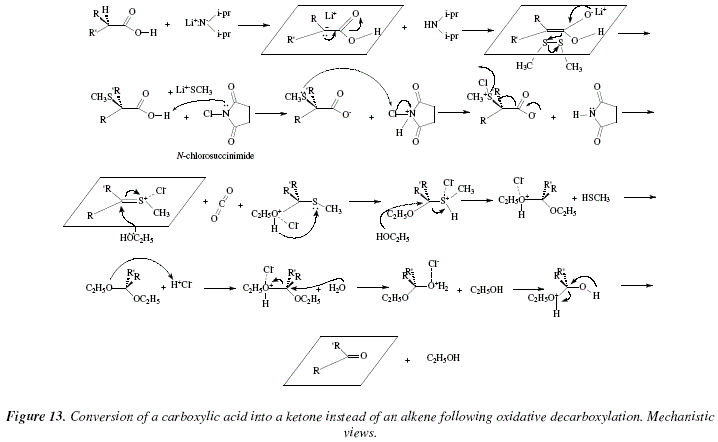
Figure 13 shows the substrate under attack by a strong base, Li+N-(i-pr)2. The proton in ![]() presents an extra acidic character due to the carbonyl group. This proton interacts with the base Li+N-(i-pr)2 to form a carbanion. The negative charge (two electrons) over the carbanion, delocalize to form the alkene's double bond and thus to dispatch the mobile
presents an extra acidic character due to the carbonyl group. This proton interacts with the base Li+N-(i-pr)2 to form a carbanion. The negative charge (two electrons) over the carbanion, delocalize to form the alkene's double bond and thus to dispatch the mobile ![]() electrons of carbonyl to the carbonyl's oxygen to generate an alcoxy group. The allylalcoxy (stabilized by the cation Li+) moves its
electrons of carbonyl to the carbonyl's oxygen to generate an alcoxy group. The allylalcoxy (stabilized by the cation Li+) moves its ![]() electrons as nucleophile over dimethyl disulphide breaking the inter-sulfur linkage, all provoked by the reformation of the carbonyl group by localization of the negative excessive charge on oxygen. The
electrons as nucleophile over dimethyl disulphide breaking the inter-sulfur linkage, all provoked by the reformation of the carbonyl group by localization of the negative excessive charge on oxygen. The ![]() -methyl thioether carboxylic acid, after the introduction of the S-group in
-methyl thioether carboxylic acid, after the introduction of the S-group in ![]() , presents now acidity enough to interact with the N-chlorosuccinimide. The N-chlorosuccinimide protonated frees a chloride (Cl-) by means of an nucleophilic attack from the sulfur from the
, presents now acidity enough to interact with the N-chlorosuccinimide. The N-chlorosuccinimide protonated frees a chloride (Cl-) by means of an nucleophilic attack from the sulfur from the ![]() -methyl thioether carboxylic acid. The chlorinated thioether carboxylate residue frees a chloride by decarboxylation and formation of the double bond between carbon and sulfur. A double nucleophilic addition of ethanol with configuration inversion occurs with this substrate to generate an ethyl-acetal which by means of protonation of the first ethanol residue and subsequent separation as alcohol by hydrolysis and elimination of the second protonated ethanol, finally conducts to the ketone product. Figure 14 is a similar mechanism to figure 13.
-methyl thioether carboxylic acid. The chlorinated thioether carboxylate residue frees a chloride by decarboxylation and formation of the double bond between carbon and sulfur. A double nucleophilic addition of ethanol with configuration inversion occurs with this substrate to generate an ethyl-acetal which by means of protonation of the first ethanol residue and subsequent separation as alcohol by hydrolysis and elimination of the second protonated ethanol, finally conducts to the ketone product. Figure 14 is a similar mechanism to figure 13.
ACKNOWLEDGEMENTS
The authors express their gratitude to Prof. Eduardo Palenque from the Department of Physics, Universidad Mayor de San Andrés, for his bibliographic support.
REFERENCES
1. Bravo, J. 2005, Rev. Bol. Quim., 23, 1. (http://www.bolivianchemistryjournal.org, 2005). [ Links ]
2. Bravo, J.A., Mollinedo, P., Peñarrieta, J.M., Vila, J.L. 2013, Rev. Bol. Quim., 30, 24 (http://www.bolivianchemistryournal.org, 2013). [ Links ]
3. Bravo, J.A., Vila, J.L. 2014, Rev. Bol. Quim., 31, 61 (http://www.bolivianchemistryournal.org, 2014). [ Links ]
4. Bravo, J.A., Vila, J.L. 2015, Rev. Bol. Quim., 32, 15 (http://www.bolivianchemistryjournal.org, 2015). [ Links ]
5. Vila, J.L., Bravo, J.A. 2015, Rev. Bol. Quim., 32, 37 (http://www.bolivianchemistryjournal.org, 2015). [ Links ]
6. Carruthers, W. Some Modern Methods of Organic Synthesis, Cambridge University Press, 3rd ed., 1987, Worcester, U.K., pp. 159-162. [ Links ]
7. Sheldon, R.A., Kochi, J.K. 1972, Organic Reactions, 19, 279. [ Links ]

