










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO  uBio
uBio
Share

 Permalink
PermalinkRevista Boliviana de Química
On-line version ISSN 0250-5460
Rev. Bol. Quim vol.32 no.1 La Paz Apr. 2015
ARTICULOS ORIGINALES
Mechanistic views of stereoselective synthesis of tri-and tetra-substituted alkenes, part II; the organic chemistry notebook series, a didacticalapproach, N° 4
José A. Bravo*, José L. Vila
Department of Chemistry, Laboratorio de Síntesis y Hemisíntesis, Instituto de Investigaciones en Productos Naturales IIPN, Universidad Mayor de San Andrés UMSA, P.O. Box 303, Calle Andrés Bello s/n, Ciudad Universitaria Cota Cota, Tel. 59122792238, La Paz, Bolivia
*Corresponding author:jabravo@umsa.bo ANALYSIS AND MECHANISTIC PROPOSALS
Abstract
This is the complementing part of the previously published: "Mechanistic views of stereoselective synthesis of tri-and tetra-substituted alkenes, part I; The Organic Chemistry Notebook Series, a Didactical Approach, N° 3". As underlined in three previous papers the presentation of synthesis works in a verbal and graphical succinct manner, needs a didactical approach. Isomerically pure tri- and tetra-substituted alkenes are difficult to obtain as shown in several publications. We used a series of reactions to synthesize tri- and tetra-substituted alkenes as reviewed by W. Carruthers, and we have proposed didactical and mechanistic views for the reviewed reactions. These two latest approaches are included in the synthetic methods reviewed by W. Carruthers with respect to the "Formation of carbon-carbon double bonds". Spanish title: Vistas mecanísticas de síntesis estereoselectivas de alquenos tri y tetrasubstituidos, Parte II, Serie: El cuaderno de notas de química orgánica, un enfoque didáctico, N°4.
Keywords: Organic Chemistry, Stereoselective synthesis, Alkenes, Addition reaction, Mechanisms of Reactions.
ANALYSIS AND MECHANISTIC PROPOSALS
As academics we are concerned with the didactical importance of covering the needs of debutant students in organic synthesis. This is the fourth study in: "The Organic Chemistry Notebook Series, a Didactical Approach" [1-3]. The present article is the completion of the analytical and didactical approach to stereoselective synthesis of tri- and tetra-substituted alkenes as reviewed by W. Carruthers [4], and illustrated mechanistically by J. Bravo and J. Vila [3]. Tri-and tetra-substituted alkenes are difficult to obtain. Authors [4] mentioned other methods signaled as useful for the stereospecific synthesis of alkenes starting with alkynes. The procedure starts with alkenylalanes and alkenylboranes. The first are obtained by hydroalumination of alkynes with e.g. diisobutylaluminium hydride. The addition is cis type, giving rise to Z-alkenylalanes. The Z-alkenylalanes react with halogens with retention of configuration to afford alkenyl halides. The halogen atoms can be replaced by alkyl groups by means of reacting with an organocopper reagent. Iodination of the alane obtained from reaction of 1-hexyne with diisobutylaluminium hydride makes possible the (E)-1-iodo-1-hexene. 3-hexyne gives (E)-3-iodo-3-hexene [4]. See Figure 1 for the reactions reviewed by W. Carruthers [4].
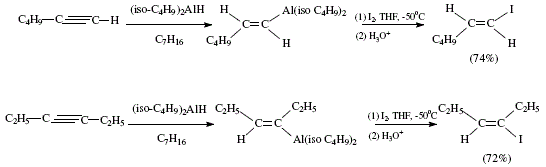
Figure 1. Obtention of alkenyl halides via alkenylalanes [(E)-1-iodo-1-hexene and (E)-3-iodo-3-hexene; reviewed by W. Carruthers [4]
Figure 2 shows the mechanistic proposal for these reactions.
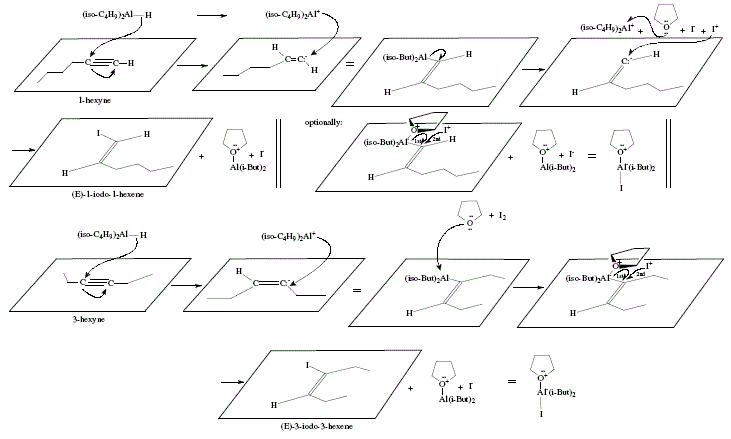
Figure 2. Synthesis of (E)-1 -iodo-1 -hexene and (E)-3-iodo-3-hexene, mechanistic views
Alternatively, Z- and E-1-halogenoalkenes can be obtained from terminal acetylenes by means of the use of E-alkenylboronic acids which can be synthesized by reaction of terminal acetylenes with catecholborane and subsequent hydrolysis [4]. Boronic acids react with sodium hydroxide to replace the boronic acid group by iodine to afford E-1-iodoalkene; retention of configuration occurs [4]. If the reaction is carried out with bromine and base, the substitution result implies inversion; Z-1-bromoalkene is obtained [4]. In both reactions, the stereoselectivity is high; for example, 1-octyne gave (Z)-1-bromo-1-octene (85% yield, 99% stereoselectivity), and 1-hexyne gave (E)-1-iodo-1-hexene [4]. Figure 3 and 4 show the global reactions and the mechanistic approach, respectively.
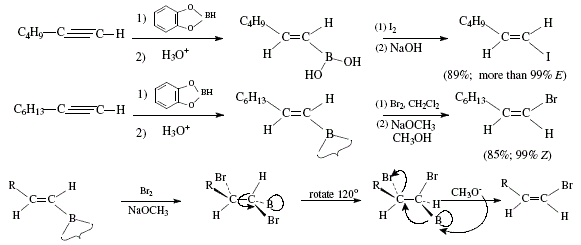
Figure 3. Obtention of (E)-1-halogenoalkenes from terminal acetylenes via E-alkenylboronic acids. Inversion of configuration
duringbromination. Reviewed by W. Carruthers [4]
Inversion of configuration in brominating is explained by the trans-addition of Br2 to the double bond and ulterior trans-elimination of boron and bromine due to NaOCH3 ([4], Figure 3).
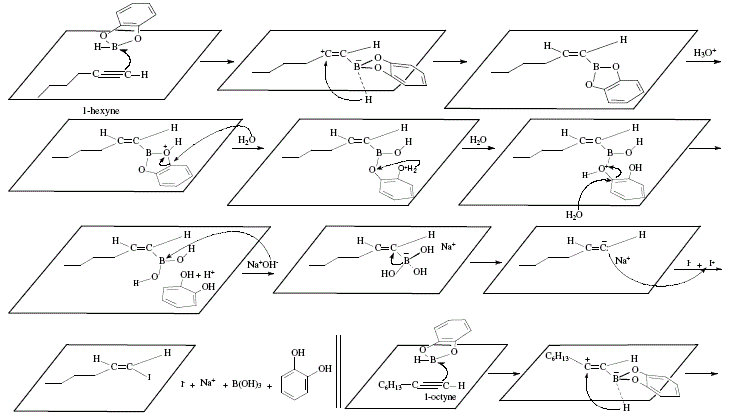
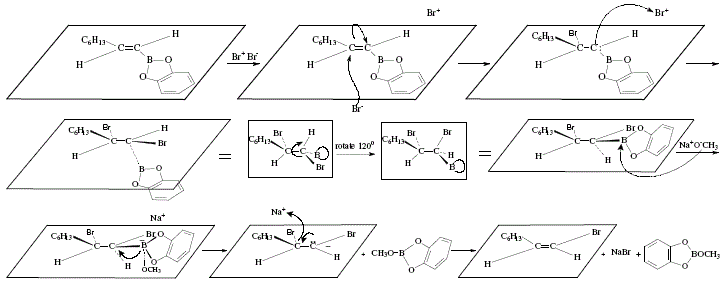
Figure 4. Obtention of (E)-1 -halogenoalkenes from terminal acetylenes via E-alkenylboronic acids. Inversion of configuration
during bromination, mechanistic view
On other topics, the reviewer mentions o^-unsaturated carboxylic acids obtained from aluminates that at their turn were afforded by alkenylalanes after reaction with methyl-lithium. In the same manner, paraformaldehyde and other similar compounds give rise to allylic alcohols [4,5]. The difference between this reaction and the one previously exposed regarding hydroalumination of disubstituted acetylenes with lithium hydridodiisobutylmethylaluminate obtained from diisobutylalumimium hydride and methyl-lithium, is the trans addition to the alkyne multiple bond [4,5]. Carbonation gives c^-unsaturated carboxylic acids. The reaction with paraformaldehyde/acetaldehyde gives allylic alcohols; iodine, alkenyl iodides. All these derivatives are isomeric with compounds obtained using diisobutylalumimium hydride discussed previously [4,6]. Both isomers cis and trans a-methylcrotonic acids were obtained from 2-butyne [4,6]. Figure 5 shows the reaction sequences; Figure 6 the applicable theoretical mechanistic proposals. 'In all these reactions the isobutyl groups of the hydroaluminating agent are converted into isobutane in the hydrolysis step, and do not interfere with the isolation of the products' (W. Carruthers [4]). See Figure 6.
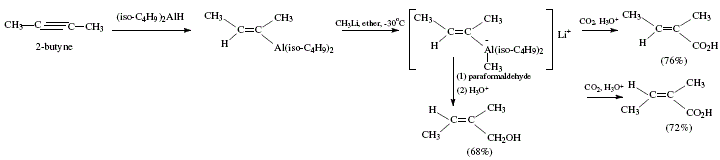
Figure 5. Obtaining of ap-unsaturated carboxylic acids and allylic alcohol from aluminates; reviewed by W. Carruthers [4]
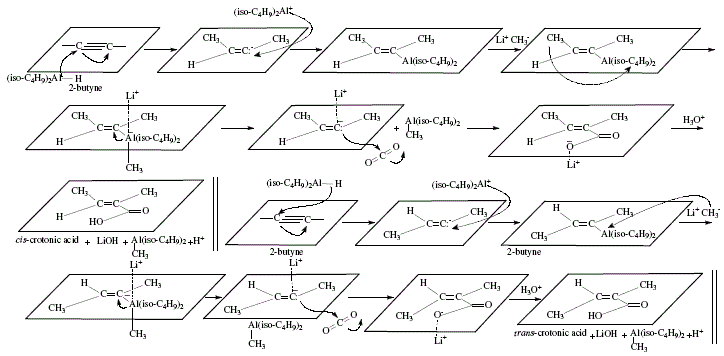
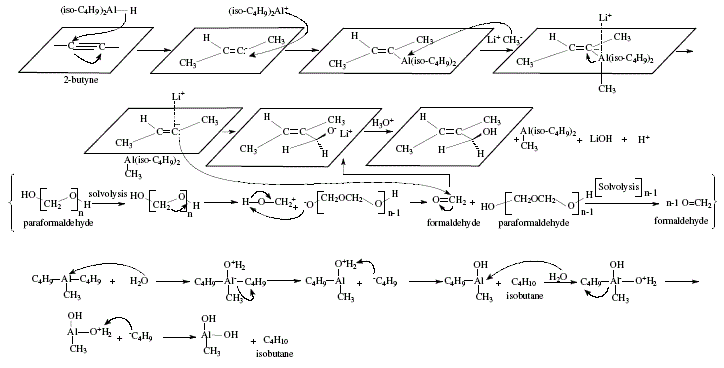
Figure 6. Obtaining of afi-unsaturated carboxylic acids and allylic alcohol from aluminates; mechanistic views
In contrast with alkenylalanes, alkenylboranes (obtainable from hydroboration of alkynes) exhibit a different comportment. Under iodination, no iodide is produced, instead, the stereospecific transfer of one alkyl group from boron to its vicinal carbon occurs. This provides an alternative stereospecific synthesis of substituted alkenes [4]. In this way, iodine and sodium hydroxide added to the alkenylborane obtained after hydroboration of 1-hexyne with dicyclohexylborane, conducts to (Z)-1-cyclohexyl-1-hexene (75% yield). See Figure 7 for the reaction reviewed by W. Carruthers [4]. Figure 8 shows the corresponding mechanistic view.
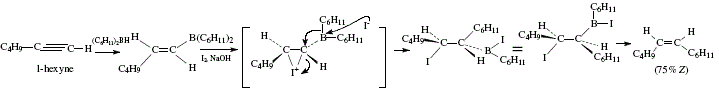
Figure 7. Synthesis of (Z)-1 -cyclohexyl-1 -hexene employing 1-hexyne as starting material and applying alkenylborane and
subsequent addition of iodine; reviewed by W. Carruthers [4]
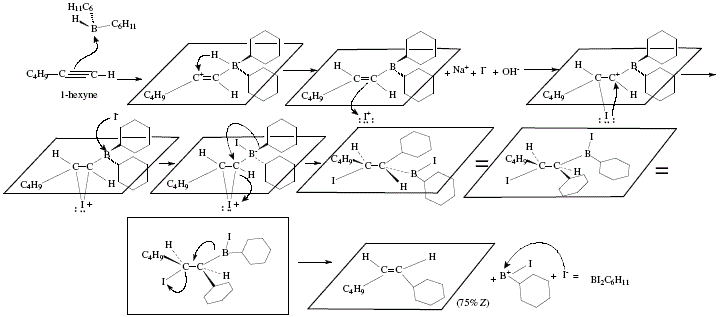
Figure 8. Synthesis of (Z)-1 -cyclohexyl-1 -hexene employing 1-hexyne as starting material and applying alkenylborane and
subsequent addition of iodine; mechanistic views
Similarly, 3-hexyne is transformed in (Z)-3-cyclohexyl-3-hexene [4]; Figure 9 shows the corresponding mechanism. The "anti elimination of boron and the iodine atom, reassures the stereoselectivity of the observed alkenic product in the alkene-forming step [4].
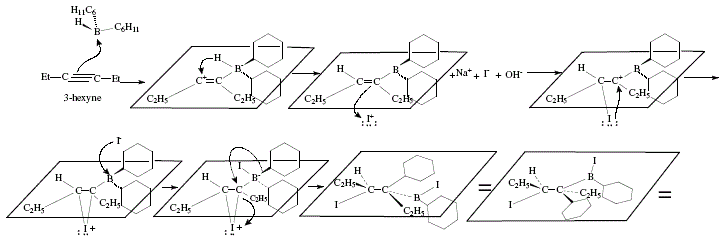
Figure 9. Synthesis of(Z)-3-cyclohexy l-3-hexene employing 3-hexyne as starting material and applying alkenylborane and
subsequent addition of iodine; mechanistic views.
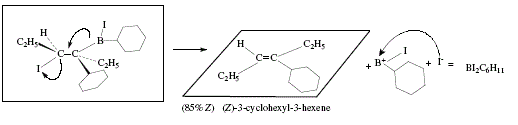
Figure 9. Synthesis of(Z)-3-cyclohexyl-3-hexene employing 3-hexyne as starting material and applying alkenylborane and
subsequent addition ofiodine; mechanistic view (Cont.)
Dialkylvinylboranes can be synthesized from dialkylhaloboranes and an internal alkyne in the presence of lithium aluminium hydride. These can react with iodine in the presence of methoxide to produce trisubstituted alkenes, stereoselectively. The two substituents of the alkyne are trans to each other [4]. (Z)-3-methyl-3-pentene was obtained from 2-butyne as Figure 10 shows [4]. The mechanism includes the iodine complex already exposed in Figures 7 to 9; see Figure 11.
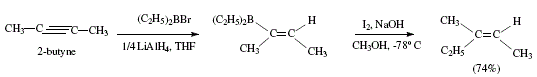
Figure 10. Synthesis of (Z)-3-methyl-2-pentene, a tri-substituted alkene with the two original substituents in alkyne located in
trans to each other; reviewed by W. Carruthers [4]
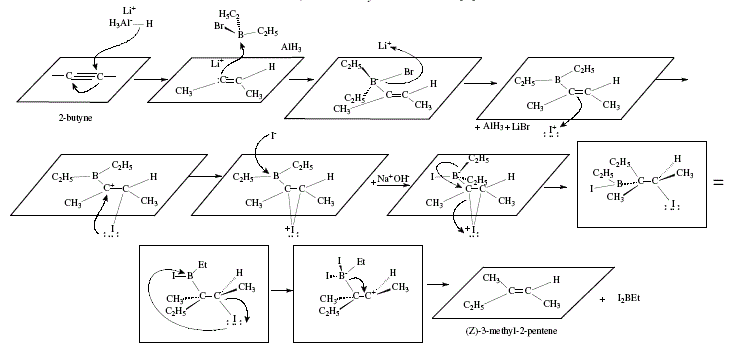
Figure 11. Synthesis of (Z)-3-methyl-2-pentene, a tri-substituted alkene with the two original substituents in alkyne located in
trans to each other; mechanistic views
Trisubstituted alkenes are also obtained from terminal alkynes by way of the corresponding 1-alkenyl iodides. Iodide is treated by butyl-lithium, trialkylborane and iodine. The trisubstituted alkene possesses the iodine of the alkenyl iodide replaced by an alkyl group coming from the borane with retention of configuration. 'The stereochemistry of the product requires syn elimination of R2BI' [4,7,8]. Figures 12 and 13 explain this synthesis.
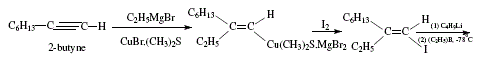
Figure 12. Trisubstituted alkene synthesis by iodination and subsequent treatment with butyl-lithium, trialkylborane and iodine;
reviewed by W. Carruthers [4]
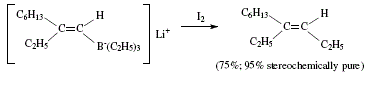
Figure 12. Trisubstituted alkene synthesis by iodination and subsequent treatment with butyl-lithium, trialkylborane and iodine;
reviewed by W. Carruthers [4] (Cont.)
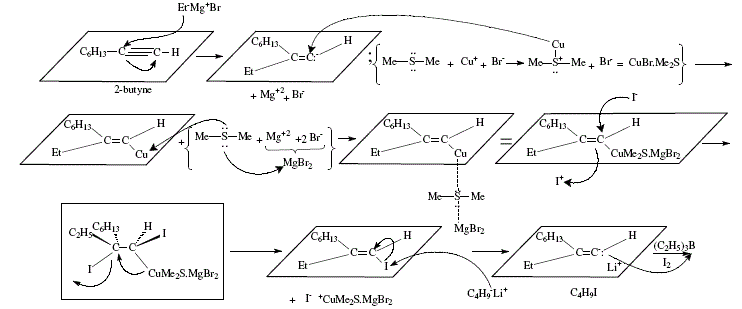
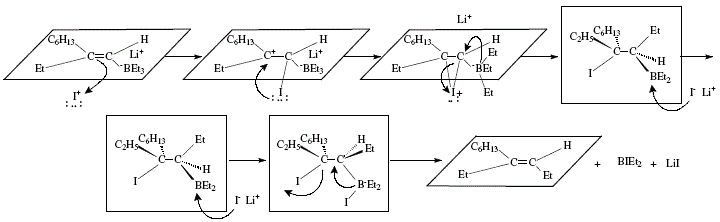
Figure 13. Trisubstituted alkene synthesis by iodination and subsequent treatment with butyl-lithium, trialkylborane and iodine;
mechanistic views
The synthesis of trisubstituted alkenes can be achieved using allylic alcohols as starting material. This depends in its critical phase on the stereoselective epoxidation of allylic alcohols. Hence, (Z)-6-methyl-5-undecene was obtained by oxidation of the allylic alcohol 1 with t-butyl hydroperoxide and vanadium catalyst to selectively give rise to erythro-epoxide 2 that at its turn was converted to the vicinal diol 3 thanks to interaction with lithium dibutylcuprate. Stereospecific deoxygenation of the diol could be possible by interaction with N,N-dimethylformamide dimethyl acetal. The dioxolane derivative obtained is then decomposed by heating and by adding acetic anhydride to give rise to the Z-alkene [4]. The vanadium-t-butyl hydroperoxide react with the C=C of allylic alcohols making possible selective epoxidations. Reaction of 1 with t-C4H9OOH in the presence of vanadium acetylacetonate as catalyst gave 2 almost exclusively [4,9,10]. See Figures 14 and 15.
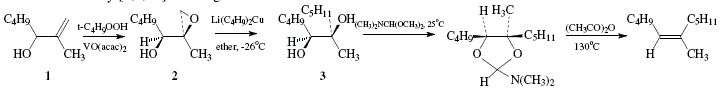
Figure 14. Trisubstituted alkene synthesis from allylic alcohol; reviewed by W. Carruthers [4]
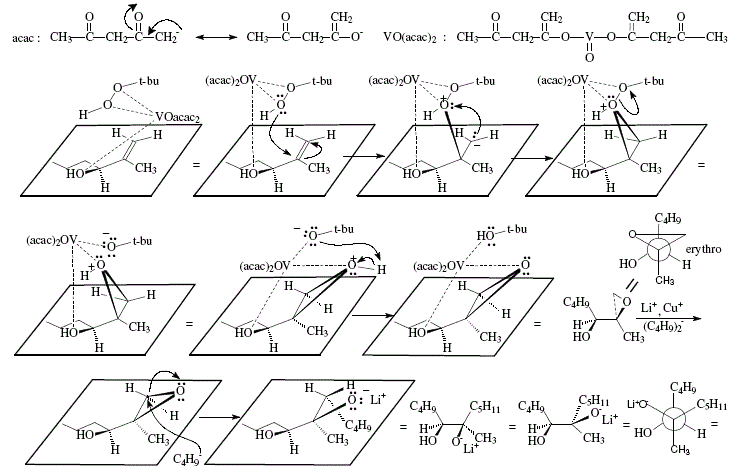
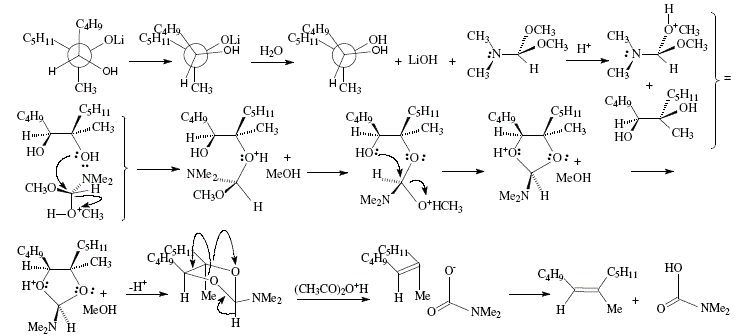
Figure 15. Trisubstituted alkene synthesis from allylic alcohol; mechanistic views
ACKNOWLEDGEMENTS
The authors express their gratitude to Prof. Eduardo Palenque from the Department of Physics, Universidad Mayor de San Andrés, for his bibliographic support.
REFERENCES
1. Bravo, J. 2005, Bol. J. of Chem., 23, 1-10. (http://www.bolivianchemistryjournal.org, 2005). [ Links ]
2. Bravo, J.A., Mollinedo, P., Peñarrieta, J.M., Vila, J.L. 2013, Bol. J. ofChem., 30, 24-41(http://www.bolivianchemistyournal.org. 2013) [ Links ]
3. Bravo, J.A., Vila, J.L. 2014, Bol. J. ofChem., 31, 61-67 (http://www.bolivianchemistyournal.or, 2014) [ Links ]
4. Carruthers, W. Some Modern Methods of Organic Synthesis, Cambridge University Press, 3rd ed., 1987, Worcester, U.K., pp. 148-156,374. [ Links ]
5. Zweifel, G., Steele, R.B. 1967, J. Am. Chem. Soc., 89, 2754. [ Links ]
6. Zweifel, G., Steele, R.B. 1967, J. Am. Chem. Soc., 89, 5085. [ Links ]
7. Lahima, N.J., Levy, A.B. 1978, J. Org. Chem., 43, 1279. [ Links ]
8. Levy, A.B., Angelastro R., Marinelli E.R. 1980, Synthesis, 845. [ Links ]
9. Sharpless, K.B., Michaelson, R.C. 1973, J. Am. Chem. Soc., 95, 6136. [ Links ]
10. Rossiter, B.E., Verhoeven, T.R., Sharpless, K.B. 1979, Tetrahedron Lett., 4733. [ Links ]

