










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO  uBio
uBio
Share

 Permalink
PermalinkRevista Boliviana de Química
On-line version ISSN 0250-5460
Rev. Bol. Quim vol.30 no.1 La Paz 2013
Artículo
FACILE SYNTHESIS OF TETRAHYDROPYRIMIDINES WITH POSSIBLEINSECTICIDAL ACTIVITY
Luis J. Reyes-García
Instituto de Química, Pontificia Universidad Católica de Valparaíso. Avda. Universidad 330.
Curauma, Valparaíso, Chile
Keywords: Tetrahydropyrimidines, insecticidal activity, heterocycles synthesis
ABSTRACT
A simple and efficient method for the synthesis of 2-(pyridin-3-yl)-1,4,5,6-tetrahydropyrimidines derivatives (THP-derivatives) from nicotinic acid was used.
RESUMEN
Se describe un simple y eficiente método para la síntesis de derivados de 2 - (3-piridinil)-1,4,5,6-tetrahidropirimidina (THP-derivados) a partir de acido nicotínico.
INTRODUCTION
Neonicotinoid insecticides (NNSs), which act as agonist on the insect nicotinic acetylcholine receptors (nAChRs) [1], these receptors are widely used targeted for insecticidal activities by different classes of tetrahydropyrimidines (THPs) [2]. By using anabasine as a témplate, several THPs were designed and prepared by the reaction of 1,3-diaminopropane and nicotinic acid, using boric acid as a catalyst (figure 1). The biological properties of which remain unexplored.
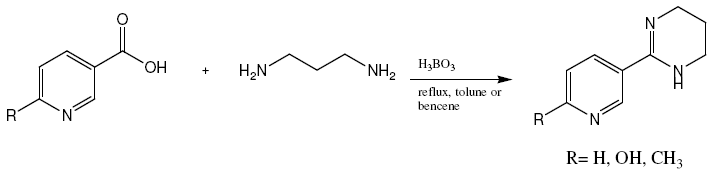
Figure 1: Synthesis of tetrahydropyrimidines via the one-pot method.
RESULTS AND DISCUSSION
The THPs were obtained by following the methodology described by Tang [3], which allows amides to be directly obtained from carboxylic acid and an amine. The obtaining of Ib was carried out through reduction of 1 with NaBH4. To obtain other THP-derivatives the same protocol in obtaining amides was used and all THP-derivatives crystallized as the hydrochloride adduct.
All the cyclizations had a low yield (less than 45%), while the yield in the reduction of 1 to obtain Ib was within the expected range, since it is known that the yield in this type of reaction is greater than or equal to 68% [4]. All The crystals obtained had the same features (white crystals), with the exception of 4 which was presented as brown crystals.
The most widely used methods for the formation of amides included the use of acid chloride in the reaction with the amine, which needed the presence of a base to neutralize the hydrochloric acid produced. Despite its wide scope, this method has several disadvantages, one being the low stability of many acid chlorides (in contact with the air are rapidly hydrolysed). Secondly, reagents used for its preparation (thionyl chloride, oxalyl chloride) are highly corrosive and toxic, even more, many functional groups present in any of the reagents must be protected to ensure the selectivity of the amide [5]. Furthermore, the thionyl chloride is convenient and economical, but is very acidic, only some acid-resistant molecules can withstand the reaction conditions. Several tests were also made using this procedure, but the results were rather disappointing (formation of undesired producís). A really effective method for obtaining amides was reported by Tang [3], in which the amide obtained by direct reaction between the amine and carboxylic acid using boric acid as catalyst.
These protocols were developed for nicotinamide analogues and then proceed to the cyclization reaction, however cyclized product was obtained directly (one-pot).
To obtain different THPs different solvents and equimolar amounts of boric acid were tested and to obtain the THP-derivatives the use of benzene and toluene was applied, with the latter solvent obtaining the best yield. With crystallization of the product, carried out in dichloromethane only nicotinic acid excess was isolated and 1 remained solid with 1,3-diaminopropane (as carbonate). To eliminate the 1,3-diaminopropane (free base), the sample was placed in a distillation equipment under reduced pressure (90 °C and 5 mmHg) after 10 h no positive results were obtained, column chromatography on silica gel was discarded because the compounds did not elute. Finally purify 1 was achieved using neutral A12O3, where the RF between compounds were quite different. The best yield for this reaction was 35% and for the THP-derivatives was 20%. This low yield can be attributed primarily to handling problems, since to obtain the puré product it must go through several stages. For example, in the case of removing excess catalyst (H3BO3) by means of washing with water alkalinized with NaOH entails a great loss of product.
The product is solubilized in water which makes extraction with dichloromethane or chloroform very difficult. This washing was not performed in the preparation of the THP-derivatives, the catalyst was removed using methanol, the solvent was then easily removed, producing an ester phase [6], however the yield was even lower than in THP. The best yield was obtained in the synthesis of 4, but the methodology used was different. It was made by melting and then using 2-propanol as a solvent to continué the reflux for a couple of hours. It is noteworthy that this procedure was attempted to obtain 1, but there was no reaction. The effect of substituent on the aromatic ring also plays an important role, because it was observed that such groups as methyl (electrón donor) favor the synthesis reducing reaction times, while electrón acceptor groups (such as chlorine) no reaction and disfavors the formation of cyclized product.
For the formation of the cyclized product monoamide and boric acid was used, it is believed that this reacts with the carboxylic acid to form a mixed anhydride, that reaction with the amine in the amide form, regenerating the catalyst [7].
CONCLUSIÓN
In summary, we have designed and synthesized a series of novel tetrahydropyrimidine analogues bearing a group in the 6 position of aromatic ring. This methodology is shown to be a good and easy alternative for the synthesis of tetrahydropyrimidines.
EXPERIMENTAL
Syntheses of compounds
Solvents and chemicals were purchased from Merck (Darmstadt, Germany) and Sigma-Aldrich (St. Louis, MO, USA). Melting points were determined with a Reichert Galen III hotplate microscope. 1H NMR spectra were recorded at 400 MHz on Bruker AMX-400 spectrometers. Chemical shifts are reported in parts per million with TMS as internal standard. Coupling constant(s) (J) in hertz, assignment. Flash chromatography was carried out on neutral alumina (70-290 mesh).
1,4,5,6-Tetrahydro-2-(pyridin-3-yl)pyrimidine (1)
To a solution of nicotinic acid (10.0 g, 81.3 mmol) in toluene (250 mL) were added B(OH)3 (0.31 g, 4.17 mmol) and 1,3-diaminopropane (6.8 mL, 81 mmol) and the mixture was boiled under reflux with a Dean-Stark trap for five days. After this time, 2.5 mL of H2O had been collected, and the solvent was removed in a rotary evaporator. The mixture was made strongly basic (pH > 12) with NaOH (2 M) and extracted with CH2Cl2 (3 x 100 mL). The extract was washed with brine, dried over sodium sulfate, and concentrated to dryness. The residue was subjected to chromatography on alumina, eluting with MeOH containing 5% (v/v) of 28% NH3 (aq.) to give 1,4,5,6-tetrahydro-2-(pyridin-3-yl)pyrimidine as a colorless oil (3.2 g, 35%). This was treated with HCl in ether and the volátiles were removed to give an off-white solid, mp 269-273 °C. 1H NMR (HCl salt; 400 MHz, D2O) ![]() 1.91 (p, 2H, J= 5.8 Hz), 3.41 (t, 4H, J= 5.7 Hz), 7.65 (ddd, 1H, J= 7.9, 4.9, 0.6 Hz), 8.20 (dt, 1H, J= 7.9, 1.9 Hz,), 8.85 (dd, 1H, J= 4.8, 1.7 Hz), 8.91 (d, 1H,J= 1.8Hz);13CNMR(D2O,
1.91 (p, 2H, J= 5.8 Hz), 3.41 (t, 4H, J= 5.7 Hz), 7.65 (ddd, 1H, J= 7.9, 4.9, 0.6 Hz), 8.20 (dt, 1H, J= 7.9, 1.9 Hz,), 8.85 (dd, 1H, J= 4.8, 1.7 Hz), 8.91 (d, 1H,J= 1.8Hz);13CNMR(D2O, ![]() ): 18.5,39.4, 124.3, 125.6, 136.7, 149,3, 154.1, 158.2
): 18.5,39.4, 124.3, 125.6, 136.7, 149,3, 154.1, 158.2
Hexahydro-2-(pyridin-3-yl)pyrimidine (1b)
NaBH4 (1.41 g, 37 mmol) was added gradually to a solution of 1,4,5,6-tetrahydro-2-(pyridin-3-yl)pyrimidine (0.4 g, 2.48 mmol) in MeOH/CHCl3 (1:1) (50 mL) and the mixture was stirred at room temperature for 24 h The reaction mixture was adjusted to pH = 1 with HC1 (2 M), stirred at room temperature for 10 min, and concentrated to dryness under reduced pressure. The residue was dissolved in CH2C12 (100 mL), made basic (pH ~ 10) with 10% aqueous NaOH and extracted with CH2C12 (2 x 50 mL). The combined extracts were washed with brine (3 x 50mL), dried over Na2SO4, and concentrated to dryness. The residue was purified by chromatography on alumina (MeOH:Et2O 4:1) to give hexahydro-2-(pyridin-3-yl)pyrimidine as a colorless oil (0.34 g, 68% yield). This was treated with HC1 in MeOH:Et2O and the volátiles were removed to give an off-white solid, mp 175-179 °C, 1H-NMR (DMSO-<i6), 5 2.05 (p, 2H, J= 7.4 Hz), 2.53-2.47 (m, 2H, ), 3.01-2.85 (m, 4H,), 4.31 (s, 1H), 7.91 (t, 1H, J= 6.6 Hz), 8.60 (d, 1H, J = 8.0 Hz), 8.86 (dd, 1H, J= 5.4, 1.3 Hz), 9.05 (d, 1H, J= 1.6 Hz); 13C NMR (DMSO-<i6, ![]() ): 19.7, 36.5, 72.6, 124.3, 134.5, 146.2, 153.9.
): 19.7, 36.5, 72.6, 124.3, 134.5, 146.2, 153.9.
1,4,5,6-Tetrahydro-2-(6-methylpyridin-3-yl)pyrimidine (2)
To a solution of 6-methylnicotinic acid (1.00 g, 7.29 mmol) in benzene (50 mL) were added B(OH)3 (0.31 g, 4.17 mmol) and 1,3-diaminopropane (0.61 mi 7.29 mmol) The mixture was stirred at room temperature for 1 h and then at reflux temperature for further 24 h MeOH was added to the reaction mixture to remove excess catalyst as B(OMe)3 and the volátiles were removed. Flash chromatography of the resulting residue on alumina using MeOH yielded 0.25 g (20%) of colorless oil. This product was dissolved in MeOH (4 mL) and HC1 (1 M in Et2O, 4 mL) was added with a syringe pump over 50 min at room temperature. After stirring for 30 min, the solvent was removed. The residue was recrystallized from MeOH-Et2O to give l,4,5,6-tetrahydro-2-(6-methylpyridin-3-yl)pyrimidine.HCl as a white solid; mp 197-199°C, 1H-NMR (D2O), 5 1.93 (q, 2H, J= 5.8 Hz), 2.70 (s,3H), 3.48 (t, 4H, J= 5.6 Hz), 7.90 (d, 1H, J= 8.1 Hz), 8.49 (dd, 1H, J= 8.1, 2.3 Hz), 8.86 (d, 1H, J= 1.9 Hz); 13C NMR (D2O, ![]() ): 18.6, 24,6, 39.6, 124.3, 125.8, 136.9, 149,5, 154.3
): 18.6, 24,6, 39.6, 124.3, 125.8, 136.9, 149,5, 154.3
5-(1,4,5,6-Tetrahydropyrimidin-2-yl)pyridin-2-ol (3)
6-Hydroxynicotinic acid (1.0 g, 7.19 mmol) and 1,3-diaminopropane (0.6 mL, 7 mmol) in toluene (50 mL) were added to a suspensión of B(OH)3(0.74 g, 10 mmol) in toluene (30 mL) and the mixture was stirred at room temperature for 1 h and then at reflux temperature for further 24 h. MeOH was added to the reaction mixture and the volátiles were removed. The residue was subjected to chromatography on alumina, eluting with MeOH containing 5% of 28% aqueous NH3 to give 5-(l,4,5,6-tetrahydropyrimidin-2-yl)pyridin-2-ol as a colorless oil (0.15 g, 11%). This was treated with HC1 in Et2O and concentrated to dryness to give a white solid; mp 297-303°C, 1H-NMR (D2O), 5 1.71 (p, 2H, J= 5.8 Hz), 3.35 (t, 4H, J= 6.9 Hz), 6.53-6.46 (m, 1H), 7.64-7.61 (m, 1H), 7.85 (s, 1H); 13C NMR (D2O, ![]() ): 18.8,40.1, 124.1, 125.9, 135.4, 146.3, 154.5, 158.6
): 18.8,40.1, 124.1, 125.9, 135.4, 146.3, 154.5, 158.6
6-(1,4,5,6-Tetrahydropyrimidin-2-yl)pyridin-2-ol (4)
To a solution of 6-hydroxypyridine-2-carboxylic acid (0.6 g, 4.31 mmol) in iPrOH (50 mL) was added 1,3-diaminopropane (0.6 mL, 7 mmol) and the mixture was stirred at room temperature for 1 h, after which the solvent was evaporated to dryness and the reaction mixture was kept at 100 °C for 24 h. The product was subjected to chromatography on alumina, eluting with MeOH containing 5% of 28% aqueous NH3 to give 6-(l,4,5,6-tetrahydropyrimidin-2-yl)pyridin-2-ol as a light yellow oil that was treated with HC1 in Et2O and concentrated to dryness to give a brown solid (0.35 g, 46 %); mp 231-234°C, 1H-RMN (DMSO-<i6), 5 1.95 (p, 2H, J = 6.1 Hz), 3.45 (t, 4H, J= 5.8 Hz), 7.13-7.00 (m, 1H), 7.58-7.50 (m, 1H), 7.89-7.80 (m,lH); 13C NMR (D2O, ![]() ): 18.5, 39.4, 106.5, 118.4, 136.2, 150.1, 155.5, 159.2
): 18.5, 39.4, 106.5, 118.4, 136.2, 150.1, 155.5, 159.2
N-(3-Aminopropyl)-6-chloropyridine-3-carboxamide (5)
To a solution of 6-chloropyridine-3-carboxylic acid (1.00 g, 6.35 mmol) in benzene (50 mL) were added 1,3-diaminepropane (0.53 g, 7 mmol) and B(OH)3 (0.74 g, 10 mmol) and the mixture was kept at reflux for five days. The solvent was removed under reduced pressure and the crude product dissolved in CH2C12, washed twice with 10 mL of brine, and dried over MgSO4. The residue was subjected to chromatography on alumina, eluting with MeOH containing 5% of 28% (aq.) NH3to give N-(3-aminopropyl)-6-chloropyridine-3-carboxamide as a colorless oil that was treated with HC1 in Et2O and concentrated to dryness to give a white solid (0.25 g, 19 %); mp 217-220°C, 1H-NMR (D2O), 5 1.91 (p, 2H, J= 5.9 Hz), 2.95 (t, 2HJ= 5.7 Hz), 3.38 (t, 2H, J= 5.7 Hz), 6.93-6.89 (m, 1H), 8.09-7.80 (m, 1H), 8.28 (1H, s); 13C NMR (D2O), ![]() 26.5, 39.3, 40.2, 125.4, 128.2, 139.3, 145.6, 154.4, 166.1
26.5, 39.3, 40.2, 125.4, 128.2, 139.3, 145.6, 154.4, 166.1
ACKNOWLEDGMENTS
The author wishes to thank Dr. Buree Cassels and Dr. Patricio Itirriaga for providing us with invaluable suggestions, not to mention the doctoral fellowship from MECESUP UCH0601.
NOTES
*Corresponding author: luis.reyes.g@mail.pucv.cl
REFERENCES
[1] Yu H. B., Qin Z. R, Dai H., Zhang X., Qin X., Wang T., Fang J. X. "Synthesis and Insecticidal Activity of JV-Substituted (1,3-Thiazole)alkyl Sulfoximine Derivatives". Journal of Agricultural and Food Chemistry. 2008, 56, pp. 11356-11360. [ Links ]
[2] Ihara M., Okajima T., Yamashita A. et al., "Crystal structures of Lymnaea stagnalisAChBP in complex with neonicotinoid insecticides imidacloprid and clothianidin ". Invertebrate Neuroscience. 2008, 8, pp. 71-81. [ Links ]
[3] Tang, P. "Boric acid catalyzed amide formation from carboxylic acids and amines: n-benzyl-4-phenylbutyramide". Organic Syntheses. 2005, 81, pp. 262-272. [ Links ]
[4] Horii Z, Iwata C, Tamura Y. "Reduction of Phthalimides with Sodium Borohydride". Journal of Organic Chemistry. 1961, 26 (7), pp. 2273-2276. [ Links ]
[5] Kelly S. E., LaCour T. G., "A One Pot Procedure for the Synthesis of a-Hydroxyamides from the Corresponding a-Hydroxyacids". Syntheticm Communications. 1992, 22, pp 859-869. [ Links ]
[6] Liu C.H., Chen B.H., Lee, D.J., Ku J.R., Tsau F. "Trimethyl Borate Regenerated from Spent Sodium Borohydride after Hydrogen Production" Industrial & Engineering Chemistry Research. 2010, 49, pp. 9864-9869. [ Links ]
[7] Ishihara, K., Ohara, S., Yamamoto, H. "3,4,5-Trifluorobenzeneboronic Acid as an Extremely Active Amidation Catalyst". Journal of Organic Chemistry. 1996, 61, pp. 4196-4197. [ Links ]

