










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO  uBio
uBio
Share

 Permalink
PermalinkRevista Boliviana de Química
On-line version ISSN 0250-5460
Rev. Bol. Quim vol.29 no.2 La Paz 2012
BÚSQUEDA DE ESTRUCTURAS SECUNDARIAS ÓPTIMAS Y SUBÓPTIMAS DE UNA CADENA DE ARN UTILIZANDO INTELIGENCIA ARTIFICIAL
Sergio Peignier*1, Heriberto Castañeta M.2
1instituto de Ciencias Aplicadas, Bio-Informática y Modelización, Universidad de Lyon, Francia
2Instituto de Investigaciones Químicas, Carrera de Ciencias Químicas, Campus Universitario de Cota Cota Edificio FCPN c. Andrés Bello y c. 27 s/n, CP 303 La Paz, Bolivia Universidad Mayor de San Andrés, La Paz, Bolivia
*Corresponding author: Sergio.peignier@insa-lyon.fr
Accepted: 12/09/12
Published: 09/12/12
ABSTRACT
Ribonucleic acid (RNA) cannot exist in the form of linear chain, this molecule folds down for achieving its structure more stable through the formation of hydrogen bridges. The description of these bridges is called, the RNA secondary structure. From this, it is possible to deduce its tertiary structure of RNA. In many cases, this tertiary structure gives to RNA its properties, then, is interesting and important to know the secondary structure of the RNA chains. Find these structures using experimental techniques (X-ray crystallography) is slow and long, therefore, it is interesting predict these structures through computational techniques of prediction such as Artificial Intelligence. In the present work has been used a Hopfield neural network to find different secondary structures stable and an unidirectional multilayer neural network trained with real biological examples, in order to choose the secondary structure more structure next to a 'biological' real structure, between suboptimal structures encountered by the Hopfield network. This work was carried out (training of the second network, verification of the ability of prediction and validation) thanks to several samples of micro-RNA of drosophilas. Through these neural networks has been predicting a RNA secondary structure close to the "real" to a micro-RNA Sosophora Drosophila willistoni.
Keywords: RNA, Secondary Structure, Neural Network, Artificial Intelligence
RESUMEN
El ácido ribonucleico (ARN) no puede existir en forma de cadena lineal, esta molécula se repliega para alcanzar su estructura más estable mediante la formación de puentes de hidrógeno. La descripción de estos puentes es llamada, estructura secundaria del ARN. A partir de ésta, es posible deducir su estructura terciaria del ARN. En muchos casos esta estructura terciaria otorga al ARN sus propiedades, entonces resulta interesante e importante conocer la estructura secundaria de las cadenas de ARN. Encontrar éstas estructuras mediante técnicas experimentales (cristalografía de rayos X) resulta lento y largo, por ende, resulta interesante predecir éstas estructuras mediante técnicas computacionales de predicción como ser Inteligencia Artificial. En el presente trabajo se ha utilizado una red neuronal de Hopfield para encontrar diferentes estructuras secundarias estables y una red neuronal multicapa unidireccional, entrenada con ejemplos biológicos reales, para elegir la estructura secundaria más próxima a una estructura "biológica" real entre las estructuras subóptimas encontradas por la red de Hopfield. Se realizó éste trabajo (entrenamiento de la segunda red, verificación de la capacidad de predicción y validación) gracias a varias muestras de micro-ARN de drosophilas. Mediante estas redes neuronales se ha logrado predecir una estructura secundaria de ARN cercana a la "real" para un micro-ARN de Drosophila Sosophora willistoni.
INTRODUCCION
El ácido Ribonucleico (ARN) tiene un rol de considerable importancia en una gran cantidad de reacciones y mecanismos necesarios para el buen funcionamiento celular [7] [8] [9] (transcripción, traducción, acciones de control, regulación, catálisis,..). El ARN está constituido por una cadena de nucleótidos unidos entre sí mediante uniones fosfodiéster. Los nucleótidos constan de tres partes: un azúcar (ribosa en el caso del ARN), una región fosfato (tri, di o monofosfato) y una base aminada. Diferentes tipos de nucleótidos constituyen el ARN, todos estos nucleótidos difieren en su base, siendo las más comunes en el ARN: la Adenida (A), el Uracilo (U), la Guanina (G) y la Citosina (C). Esta molécula no puede existir en forma de cadena lineal (estructura primaria). En efecto, esta molécula se repliega para alcanzar su estructura más estable, es decir, la estructura que presenta un nivel de energía mínima (entalpia libre de la molécula). Este repliegue se realiza gracias a la formación de puentes de hidrógeno entre las bases de los diferentes nucleótidos de la cadena de ARN. Los puentes de hidrógeno solamente se forman entre las siguientes bases: A-U (Adenina-Uracilo), C-G (citosina-guanina) y G-U (guanina-uracilo), denominadas bases complementarias de Watson-Crick. Los enlaces entre bases A-U y U-G implican la formación de dos puentes de hidrógeno y los enlaces C-G implican la formación de tres puentes de hidrógeno. Además, este repliegue depende de la temperatura y de los iones presentes en el medio acoso en el cual se encuentra el ARN [10]. Para formar un enlace, las bases deben estar separadas al menos por cinco nucleótidos; de lo contrario la estructura es inestable. Se llama estructura secundaria a la descripción de los diferentes enlaces internos de la molécula de ARN. A partir de la estructura secundaria de una molécula de ARN es posible deducir su estructura terciaria [3]. La estructura terciaria es la estructura espacial (3D) de la molécula de ARN replegada. En muchos casos esta estructura terciaria otorga al ARN sus propiedades [4] [5] [6], entonces resulta interesante e importante conocer la estructura secundaria de las cadenas de ARN.
El método más exacto para determinar la estructura secundaria de una cadena de ARN es mediante cristalografía de rayos X, sin embargo, este método físico y experimental es lento y costoso. Cada base de la cadena puede formar un puente de hidrógeno con cualquier otra base complementaria de la cadena (bases complementarias de Watson-Crick). Por ende, para una cadena de ARN existen una multitud de estructuras secundarias posibles. Todas estas se obtienen por combinatoria de puentes de hidrógeno posibles. Se podría pensar en encontrar todas estas estructuras, calcular sus energías y escoger la más estable. Sin embargo el tiempo de cálculo necesario para realizar este trabajo sería demasiado largo. Este tipo de problemas que no pueden ser resueltos en tiempo real son llamados nP completos. Existen otros métodos para predecir la estructura secundaria de una molécula de ARN, entre ellos, se encuentra el método computacional. Los primeros algoritmos capaces de predecir estructuras secundarias de cadenas de ARN fueron algoritmos de tipo dinámicos, creados por Waterman [17], Waterman y Smith [18], Nussinov [14]. Estos algoritmos son relativamente lentos y no pueden ser empleados cuando la cadena de ARN considerada es demasiado larga. Algunos algoritmos dinámicos más recientes tienen un tiempo de cálculo menor y pueden ser empleados en cadenas más largas; es el caso del algoritmo desarrollado por Zucker [1]. Existen también métodos comparativos para encontrar la estructura secundaria de una cadena de ARN. Este tipo de algoritmos trabajan simultáneamente con varias cadenas identificando estructura idénticas en cada una de ellas; es el caso del algoritmo desarrollado por Sankoff [19]. Finalmente, es posible emplear inteligencia artificial, y más precisamente redes neuronales para resolver este problema. Takefuji [13] utilizó una red neuronal de Hopfield para encontrar estructuras secundarias sub óptimas de una cadena de ARN. Otros autores como Steeg [15] [16], emplearon redes de Hopfield y máquinas de Boltzman para predecir estas estructuras. Sin embargo, muchos autores asumen que la estructura más estable (mínimo energético) es la estructura biológica de la cadena de ARN en cuestión. Sin embargo, en el medio biológico en el cual se encuentra la molécula de ARN podrían existir factores que actúen sobre la molécula de ARN para que ésta adquiera otra estructura estable (sub óptima) diferente de la óptima. En este caso, la estructura biológica también podría ser diferente de la que corresponde a la estructura óptima. En el presente trabajo de investigación se ha utilizado una red neuronal de Hopfield (como en el trabajo de Takefuji [13]) para encontrar diferentes estructuras secundarias estables, (correspondientes a mínimos locales y global de energía) de una cadena de ARN. Posteriormente se utilizó una red neuronal multicapa unidireccional, entrenada con ejemplos reales para predecir la estructura biológica. A continuación se describen algunas definiciones necesarias así como los descriptores que fueron utilizados para caracterizar una estructura secundaria de ARN.
Definiciones y descripción de los descriptores de una estructura secundaria de ARN
Los nucleótidos de la cadena se enumeran a partir del último nucleótido. Los puentes de hidrógeno entre las bases son llamados puentes externos, mientras que las interacciones phosphodiester son llamados puentes internos [1]. Imaginemos la formación de dos puentes externos: imaginemos que los nucleótidos en posiciones i y j pueden formar pueden formar un puente externo y que los nucleótidos en posiciones i' y j' pueden formar otro puente externo. Estos dos puentes posibles son contradictorios en los casos siguientes:
- Si estos puentes se cruzan". Es decir si i < i' <j <j' o si i' < i <j' <j.
- Si dos puentes posibles involucran a un mismo nucleótido. Es decir si i=j' o j=i' o i=i' o j=j'.
- Si dos puentes posibles son contradictorios entonces solo uno de esos enlaces posibles podrá existir.
Toda región totalmente rodeada por puentes es llamada "figura" [1]. Existen cinco clases de "figuras" distintas. Cada una de estas clases se encuentra en la tabla 1 y en la figura 2.

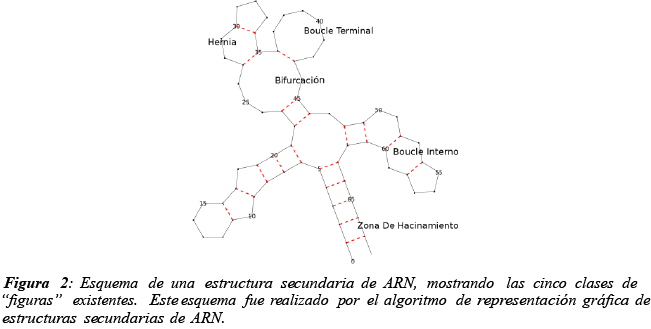
Las bifurcaciones marcan el nacimiento de "brazos" en la estructura secundaria. Los "brazos" nacen de los puentes externos de la bifurcación. La energía asociada a una estructura secundaria es igual a la suma de las energías asociadas a cada una de las figuras que la componen [1]. Estas energías son datos termodinámicos establecidos experimentalmente [11]. La estabilidad de la estructura secundaria es una medida que está ligada a la energía total de la estructura secundaria de ARN. La estabilidad depende del número de puentes de hidrógeno presentes en la estructura secundaria de ARN y del tipo de bases que los forman. En este trabajo se decidió utilizar la estabilidad en lugar de la energía para poder caracterizar una estructura secundaria de ARN, ya que el cálculo de la estabilidad no depende de ninguna tabla y se realiza fácilmente. Sin embargo, sería relativamente sencillo calcular la energía de la estructura secundaria gracias a las tablas ya mencionadas [11]. El porcentaje GC corresponde al número total de puentes de hidrógeno entre una base C y una base G dividido por el número total de puentes de hidrógeno de la estructura secundaria de ARN. Ya se mencionó en la introducción que un enlace C-G implica la formación de 3 puentes hidrógeno mientras que un enlace A-U o G-U implica la formación de 2 puentes hidrógeno. Por ende una estructura que presente un mayor porcentaje GC será globalmente más estable. Para caracterizar una estructura secundaria de ARN se utilizaron los descriptores siguientes:
- Número de figuras de Boucle terminal.
- Número de figuras de Hernia.
- Número de figuras Bifurcación.
- Número de figuras región de hacinamiento.
- Número de brazos.
- Número de puentes de hidrógeno
- Estabilidad de la estructura.
- Porcentaje de GC.
Ahora que fueron presentadas todas las definiciones necesarias y todos los descriptores de una estructura secundaria de ARN, se describirá el algoritmo empleado para predecir estas estructuras.
Algoritmo utilizado
Para realizar la predicción de estas estructuras fue necesaria la intervención de 2 redes neuronales distintas. La primera, una red neuronal de Hopfield, fue empleada para predecir un conjunto de estructuras secundarias subóptimas para una cadena de ARN. La segunda, red neuronal multicapa unidireccional, fue entrenada con ejemplos biológicos reales, y fue empleada para elegir la estructura secundaria más próxima a una estructura "biológica" real entre las estructuras subóptimas encontradas por la primera red neuronal.
Redes neuronales
En ambos casos fueron empleadas redes neuronales. Este tipo de estructuras de Inteligencia Artificial nacen como simplificación de las redes neuronales existentes en los sistemas nerviosos animales (como por ejemplo, el cerebro humano). Las neuronas son células constituidas por tres regiones principales, las dendritas o conexiones entrantes, el cuerpo celular o soma y una conexión de salida o axona. La señal nerviosa se desplaza desde las dendritas hasta el axona pasando por el soma. En función de las señales recibidas (inhibición o excitación), la neurona producirá o no una señal nerviosa de salida. Esta señal llegará a su vez a las dendritas de otras neuronas y así sucesivamente, cubriendo toda la red neuronal y pudiendo llegar a elementos motores (músculos ...). Una neurona puede modelizarse mediante los elementos siguientes:
- Un peso asociado a cada elemento del vector de entrada (modelización de una acción de excitación o inhibición sobre la neurona).
- Una función de suma ponderada de los elementos de entrada y sus pesos asociados.
- Un valor límite o "umbral": si la suma ponderada está por debajo de este valor, no habrá respuesta por parte de la neurona; de lo contrario, si la suma ponderada es superior, entonces la neurona responderá al vector de entrada.
- Un valor de salida o función de activación que modeliza la respuesta neuronal (axona).
Las redes neuronales empleadas en este trabajo tienen una organización similar a la expuesta. Sin embargo, estas redes neuronales poseen algunas características propias que son expuestas a continuación:
Algoritmo de búsqueda de estructuras secundarias subóptimas de ARN: Red neuronal de Hopfield
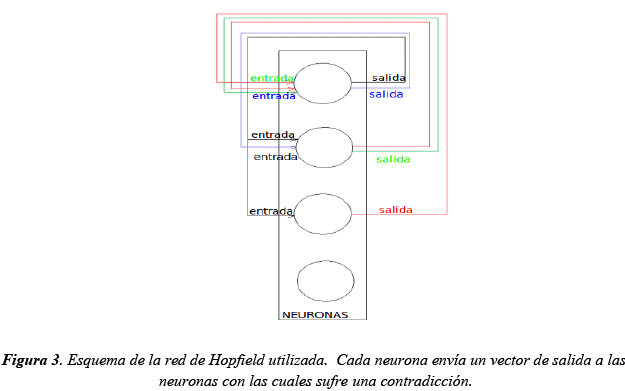
Antes de presentar el algoritmo utilizado, se presentan las características principales de una red neuronal de Hopfield. Una red de Hopfield está compuesta por N neuronas, cada una de ellas está conectada con las demás neuronas; pero no con ella misma. Es decir, que cada neurona recibe como vector de entrada todas las funciones de activación de las demás neuronas de la red. Este tipo de redes neuronales son utilizadas principalmente para memorizar patrones (imágenes...) o para procesos de optimización. En este trabajo fue utilizada con este último objetivo: optimizar la estabilidad de una estructura secundaria de ARN (encontrar las estructuras subóptimas). Cada neurona representa un puente externo posible entre dos nucleótidos de la cadena de ARN. Cada una de las neuronas recibe la función de activación de las neuronas que representan otros puentes externos posibles contradictorios. De esta manera se unen entre si todas las neuronas que modelizan enlaces puentes posibles contradictorios. De modo que la función de activación de cada neurona funciona como un "foco". Es decir que si el "foco" está prendido (si la neurona esta activada) significa que el puente representado fue "elegido" entre los demás puentes contradictorios cuyos "focos" estarán apagados.
La ecuación diferencial que esta detrás de la evolución temporal de la función de activación de la i- ésima neurona (brillo del "foco") es la siguiente:
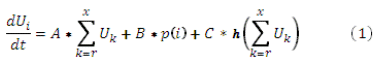
Donde: Ui es la función de la i-ésima neurona, ![]() corresponde a la derivada de la función de activación de la neurona,
corresponde a la derivada de la función de activación de la neurona, ![]() es la suma de las funciones de activación de las demás neuronas contradictorias. Este término inhibe la evolución de la función de activación (baja la luminosidad del "foco"), P(i) corresponde a la energía del puente representado por la neurona en cuestión (1 si se trata de de A-U y 2 si se trata de C-G [12]) Este término favorecerá la evolución de la función de activación (aumenta la luminosidad del "foco"), es la función "hill-climber" (trepador de colinas) que premia a las neuronas que no poseen contradicciones, favorecerá la evolución de la función de activación (aumenta la luminosidad del "foco")(
es la suma de las funciones de activación de las demás neuronas contradictorias. Este término inhibe la evolución de la función de activación (baja la luminosidad del "foco"), P(i) corresponde a la energía del puente representado por la neurona en cuestión (1 si se trata de de A-U y 2 si se trata de C-G [12]) Este término favorecerá la evolución de la función de activación (aumenta la luminosidad del "foco"), es la función "hill-climber" (trepador de colinas) que premia a las neuronas que no poseen contradicciones, favorecerá la evolución de la función de activación (aumenta la luminosidad del "foco")(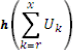 =1 si Uk = 0 es decir si no existen contradicciones, de lo contrario =0). Finalmente A, B, C son constantes. En este caso estas constantes poseen los valores siguientes: A = 0.02, B = 0.5, C = 0.03
=1 si Uk = 0 es decir si no existen contradicciones, de lo contrario =0). Finalmente A, B, C son constantes. En este caso estas constantes poseen los valores siguientes: A = 0.02, B = 0.5, C = 0.03
Una vez que se ha definido esta ecuación (1), se procede a elegir una neurona al azar y se la hace evolucionar. Se calcula el valor de su derivada (ecuación diferencial) y se modifica el valor de su función de activación. Se realiza esta acción n veces. Después de hacer evolucionar el sistema durante "n" iteraciones se puede considerar que el sistema ha logrado evolucionar hacia cierto estado subóptimo (la convergencia depende del número de iteraciones de evolución. A mayor iteración, mejor será la convergencia...). Ejecutando este algoritmo repetidas veces, se puede llegar a obtener diferentes estructuras secundarias subóptimas, más o menos distintas entre sí.
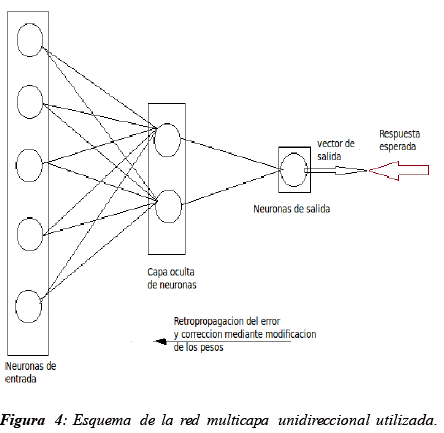
Algoritmo de selección de la estructura secundaria más próxima a una estructura "biológica ": red neuronal multicapa unidireccional
Antes de presentar el algoritmo utilizado, presentaremos las características principales de una red neuronal multi-capa unidireccional. Una red neuronal multicapa unidireccional está compuesta por N neuronas organizadas en capas. La red posee n capas y cada capa contiene Nn neuronas. La neurona de la capa i recibe como vector de entrada todas las funciones de activación de las neuronas de la capa i - 1 y envía su función de activación a todas las neuronas de la capa i + 1. Cada neurona asocia a su vector de entrada un vector de pesos propios, por ejemplo, la neurona j de la capa i asocia el vector de pesos Wi,j a su vector de entrada. Este tipo de redes neuronales son utilizadas principalmente para modelizar una relación lineal o no lineal entre dos conjuntos de datos, para procesos de optimización y para predicciones. Se la empleará para predecir la estructura secundaria más próxima a una estructura secundaria real "biológica", se establece un modelo entre los descriptores de una estructura secundaria de ARN y el carácter "biológico" de esta estructura. La red utilizada consta de tres capas de neuronas. Una capa de entrada de datos, una capa oculta y la capa de salida. La primera tiene nueve neuronas, cada una recibe uno de los descriptores de la estructura secundaria. La segunda capa presenta cinco neuronas. La última capa una única neurona. Se utilizó una función tangente hiperbólica como función de activación de las neuronas. Se tomó esta función por estar comprendida entre -1 y 1 y por ser una función sigmoidea usualmente utilizada para estos propósitos. La red fue entrenada para dar un resultado próximo a 1, si la estructura secundaria posee las características de una estructura secundaria "biológica" y 0, si no es el caso. El entrenamiento se realizó en base a estructuras secundarias de micro RNA de drosophilas. Estos datos fueron separados en tres conjuntos, un conjunto para el entrenamiento de la red, uno para verificar el poder de predicción de la red y otro para la validación del trabajo. Se consideró que la predicción podría realizarse únicamente para especies cercanas filogenéticamente a las especies de drosophilas utilizadas y para tipos de ARN similares a los utilizados para el entrenamiento (micro ARN). Utilizar este programa de predicción con tipos de ARN y especies muy lejanos no solo no tendría mucho sentido, sino podría resultar aberrante! Es claro que las características de una estructura secundaria serán distintas de un tipo de ARN a otro y de una especie a otra. El entrenamiento de la red se hizo mediante el algoritmo Back Propagation. Este Algoritmo modifica los pesos de la red neuronal con el fin de minimizar el error de la red, es decir, la distancia entre el vector de salida de la red y el resultado esperado. Para poder realizar esta minimización de error se procede a:
- Presentar sucesivamente un vector de entrada.
- La red neuronal genera un vector de salida como respuesta al vector recibido.
- Se calcula el error de la red, es decir el promedio del cuadrado de la diferencia entre cada componente del vector de salida de la red y el vector respuesta esperado.
- Se ejecuta el algoritmo Back Propagation que modifica los pesos de las neuronas de cada capa.
En el caso de los pesos de las neuronas de salida la modificación es la siguiente:
![]()
Donde:
Wi,j es el peso que le asocia la i-ésima neurona de la capa de salida a la j-ésima neurona de la capa oculta, Uouti corresponde a la señal de salida (de activación) de la i-ésima neurona de salida, λ corresponde a la tasa de aprendizaje de la neurona. El valor de esta constante determina la velocidad de convergencia del sistema. Sin embargo, si este valor es demasiado grande el sistema no converge y la red no aprende, f corresponde a la derivada de la función tangente hiperbólica utilizada como función de activación, Ansi corresponde a la respuesta esperada de la i-ésima neurona de salida y Uhidj corresponde a la señal de activación de la j - ésima neurona de la capa oculta. En el caso de los pesos de las neuronas ocultas la modificación la modificaciones la siguiente:
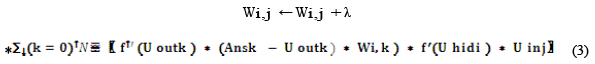
Donde:
Wi,j es el peso que le asocia la i- ésima neurona de la capa oculta a la j - ésima neurona de la capa de entrada, U inj corresponde a la señal de activación de la j - ésima neurona de la capa de entrada y Wi,k corresponde al peso que asocia la k-ésima neurona de salida a la i-ésima neurona oculta. Al repetir este proceso para cada vector de entrada, se minimiza el error de entrenamiento de la red. Sin embargo, si el tiempo de entrenamiento es demasiado largo, se corre el riesgo de sobreentrenar la red perdiendo así la capacidad de predicción y la robustez de la red. Es por esto que durante el entrenamiento se proporciona a la red un vector de entrada diferente de los vectores de entrenamiento y se calcula el error de la red. Este corresponde al error de predicción de la red. Se detiene el entrenamiento una vez que el error de entrenamiento ha bajado lo suficiente y el error de predicción no ha aumentado demasiado (sobreentrenamiento). Una vez realizado el entrenamiento, se guardan los pesos de las neuronas entrenadas para poder utilizarlos en la predicción de estructuras secundarias.
RESULTADOS Y DISCUSIÓN
Al ejecutar el primer algoritmo varias veces (para una misma cadena lineal de ARN) se obtienen varias estructuras secundarias estables (subóptimas y óptimas). Mas veces se ejecute el algoritmo, mayor es la probabilidad de encontrar la estructura secundaria óptima. Al ejecutar el algoritmo muchas veces, uno puede encontrar estructuras redundantes (una misma estructura con pequeñas variaciones) y el tiempo de cálculo crece proporcionalmente. Se decidió arbitrariamente ejecutar este algoritmo hasta obtener 100 estructuras secundarias diferentes. A continuación se muestran algunas estructuras secundarias obtenidas para el micro ARN miR-309 de la especie Drosophila Sophophora melagogaster [26], procedente de la familia de genes MIPF0000140; mir-3 en el cromosoma 2R con coordenadas: 15,547,211-15,551,279. Cuya secuencia es:
![]()
Con el fin de realizar el entrenamiento y la validación de predicción de la segunda red neuronal se utilizaron las estructuras secundarias de los micro-ARN siguientes:
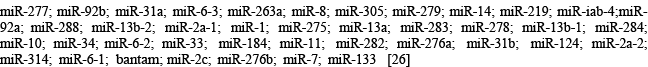
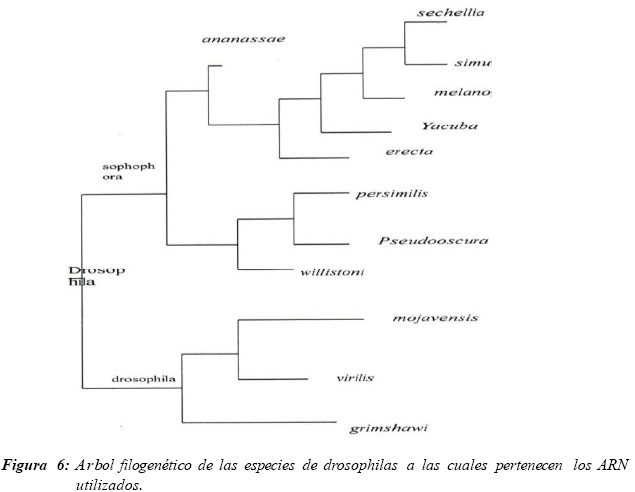
Estos micro-ARNs pertenecen a diferentes especies de Drosophilas, cuyo árbol filogenético [21] [22] [23] [24] [25] está representado en la figura 6. En un primer intento, la red fue entrenada para emitir un cierto valor (entre -1 y 1) para un tipo de ARN en particular y para una especie en particular. Pero al tener un número muy elevado de especies y de tipos de ARN para un intervalo de respuesta muy estrecho, la respuesta de la red no era satisfactoria. Por ende se decidió aumentar el número de neuronas de salida. Se creó el mismo número de neuronas de salida que de tipos de ARN. Entonces se intentó entrenar la red para que ésta active la neurona de salida correspondiente al tipo de ARN ingresado, el valor de activación dependería de la especie. Sin embargo, una vez más el aprendizaje de la red fue insuficiente. La red no logró discriminar los diferentes tipos de ARN ni las especies a las cuales pertenecen. Los descriptores correspondientes a los diferentes tipos y a las diferentes especies no son significativamente diferentes entre sí para la red neuronal. Esto puede estar ligado a tres factores. En primer lugar, las especies utilizadas son bastante cercanas, en segundo lugar, todos los ARN utilizados son micro-ARN (mismo tipo) y en tercer lugar, el número de descriptores es bastante reducido.
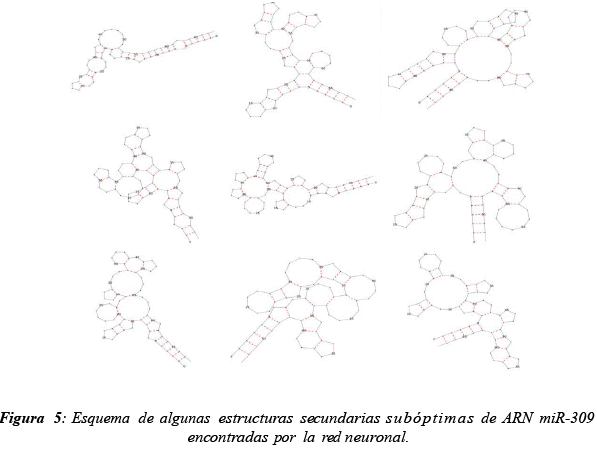
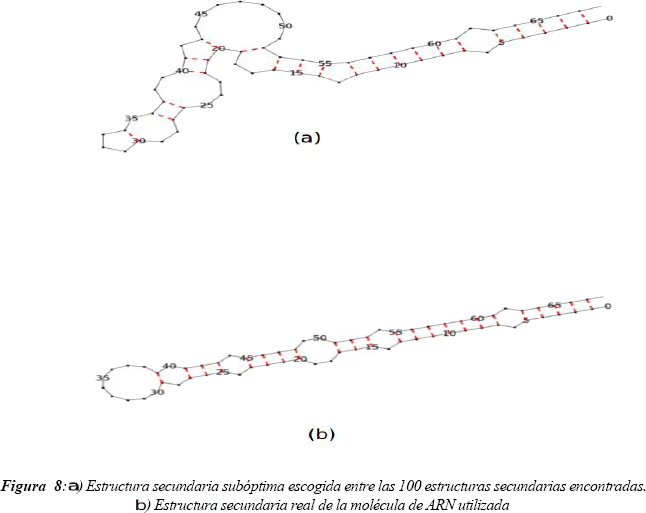
Para lograr discriminar diferentes tipos de ARN y pertenencia a diferentes especies se podría incluir nuevos descriptores para las estructuras secundarias de ARN. Esta adaptación podría ser implementada en el futuro. Sin embargo, al no ser significativamente diferentes, se consideró el conjunto de datos como uno solo, sin preocuparse de las diferencias ligadas a los tipos de micro-ARN o a las especies a las cuales pertenecen. La red fue entrenada para dar un resultado próximo a 1 si la estructura secundaria posee las características de una estructura secundaria "biológica" y 0 si no es el caso. De esta manera se repartió el conjunto de datos en tres grupos: entrenamiento, predicción y validación. Se utilizaron los vectores de descriptores de las estructuras secundarias de Drosophila Sosophora willistoni y el mismo número de "vectores de descriptores aleatorios" que no corresponden a ninguna estructura secundaria. Se incluyen estos vectores aleatorios para enseñarle a la red ejemplos de datos "falsos". De lo contrario la red no aprende a discriminar estructuras secundarias "biológicas" de "no biológicas" y aprende a afirmar que toda estructura secundaria es "biológica". Todos los otros vectores de descriptores de las demás especies, así como 10 vectores aleatorios, por cada vector de descriptores fueron utilizados como conjunto de predicción. La figura 7 ilustra la evolución de los errores de predicción y de entrenamiento de la red. Se puede ver que ambos errores disminuyen y no existe un problema de sobreentrenamiento de la red. Por tanto esta red es bastante robusta para micro ARN de drosophilas. Al predecir la estructura "biológica" del micro ARN miR-309 de la especie Drosophila Sophophora melagogaster [26] entre las 100 estructuras encontradas se encontró una estructura secundaria bastante cercana a la estructura real [26] de la molécula de ARN (figura 8)
CONCLUSIONES
El algoritmo de búsqueda de estructuras subóptimas encuentra estructuras subóptimas bastante distintas entre sí, garantizando una buena exploración del espacio de estructuras secundarias subóptimas de una molécula de ARN. Asimismo, la red neuronal entrenada para escoger la estructura más parecida a una estructura secundaria "real" elige la estructura más parecida a la real dentro de un conjunto de estructuras secundarias (las 100 estructuras encontradas por la red neuronal de Hopfield...). Se ha logrado predecir una estructura secundaria de ARN cercana a la "real" mediante la red neuronal diseñada. Sin embargo, aunque la estructura escogida se parece bastante a la estructura real solo la mitad de los puentes hidrógenos son los mismos. En efecto, la primera red neuronal explora todo el espacio de estructuras secundarias estables posibles, por ende queda atrapado en cada mínimo local existente. Ahora bien, cuanto más grande es la cadena de ARN, más combinaciones posibles de puentes de hidrógeno existen y más mínimos locales se hacen presentes. De esta manera sería necesario ejecutar muchas más veces el algoritmo de búsqueda de la red Neuronal de Hopfield para tener mayores posibilidades de encontrar a la estructura real. Sin embargo, por el tiempo de cálculo, resulta desagradable tener que ejecutar demasiadas veces este algoritmo. Para resolver este problema se podría utilizar una maquina de Boltzmann [15] en lugar de una red neuronal de Hopfield. Esto le permitiría a la red escapar de algunos mínimos locales de menor importancia y concentrarse en los mínimos locales más importantes. Al tener un conjunto de estructuras secundarias muy similares entre sí (en el caso de reemplazar una red de Hopfield por una máquina de Boltzman) se podría requerir una mayor cantidad de descriptores de la estructura secundaria de ARN, tales como potenciales electroestáticos de Markov como nuevos descriptores de la estructura secundaria de ARN [20].
REFERENCIAS
[1] ZUCKER, Michael; STIEGLER, Patrick. Optimal computer folding of large RNA sequencess using thermodynamics and auxiliary information. Nucleic Acids Research , 1981, vol. 9, no. 1
[2] MATHEWS, David; SABINA, Jeffrey; ZUCKER Michael, Expanded Sequence Dependence of Thermodynamic Parameters Improves Prediction of RNA Secondary Structure J. Mol. Biol., 1999, no. 199.2700
[3] QASIM, Romasa; KAUSER, Nishat; JILANI Tahseen,Secondary Structure prediction of RNA using Machine Learning Method International Journal of Computer Application , 2010, vol. 10, no. 6
[4] De Smit MH, van Duin J., Control of translation by mRNA secondary structure in Escherichia coli. A quantitative analysis of literatee data. J. Mol. Biol. 1994, 244(2): 144-50 [ Links ]
[5] De Smit MH, van Duin J., Secondary structure ofthe ribosomebinding site determines translational efficiency: a quantitative analysis. Proceedings of the National Academy of Sciences 1990: 7668-7672
[6] Koev G, Liu S, Beckett R, Miller WA, The 3[U+201F]-terminal structure required for replication of barley yellow dwarf viras RNA contains and embedded 3[U+201F]end, Virology 2002, 292:114-126 [ Links ]
[7] Eddy S. R. (2001) Non-coding RNA genes and the modern RNA world, Nature Rev. Genet, 2, 919-929
[8] Storz, G. (2002) An expanding universe of noncoding RNAs, Science, 296, 1260-1263
[9] Schlick, T. (2002) Molecular Modeling and Simulation: An Interdisciplinary Guide, Springer-Verlag, New York, NY.
[10] E. Westhofand P. Augginger, RNA Tertiary Structure, Encyclopediaof Analytical Chemistry, John Wiley & Songs Ltd. [ Links ]
[11]David H. Mathews, Jeffrey Sabina , Michael Zuker and Douglas H. Turner, (1999) Expanded Sequence Dependence of Thermodynamic Parameters Improves Prediction of RNA Secondary Structure, Article No. jmbi. 1999.2700
[12] Tinoco I, Uhlenbeck OC, Levine MD (1971) Estimation of secondary structure in Ribonucleic Acids. Nature 230
[13] Takefuji Y., Lin C. W., Lee K.C. (1990) A Parallel Algorithm for Estimating the Secondary Structure in Ribonucleic Acids.Biological Cybernetics
[14] Nussinov, R. and Jacobson, A. (1980). Fast Algorithm for Predicting the Secondary Structure of Single-stranded RNA.Proceedings National Academy of Sciences, U.S.A., 77:6309-6313.
[15] Steeg, E. W. (1989). Neural Network Algorithms for RNA Secondary Structure Prediction. Master's thesis, University of Toronto Computer Science Dept.
[16] Steeg, E. W. (1990). Neural Network Algorithms for RNA Secondary Structure Prediction. Technical Report CRG-TR-90-4, University of Toronto Computer Science Dept., Toronto, Canada.
[17] Waterman, M. S. (1978). Advances in Mathematics: Supplementary Studies Vol. I, Studies in Foundations and Combinatorics. Academic Press, New York.
[18] Waterman, M. S. and Smith, T. F. (1978). RNA Secondary Structure: A Complete Mathe-matical Analysis. Mathematical Biosciences, 42:257-266.
[19] Sankoff, D., Kruskal J.B., Mainville, S., and Cedergren, R. J. (1983). Fast Algorithms to Determine RNA Secondary Structures Containing Múltiple Loops. In Sankoff, D. and Kruskal, J. B., editors, Time Warps, String Edits, and Macromolecules: The Theory and Practice of Sequence Comparison. Addison-Wesley, Reading, MA.
[20] González-Díaz H., Pérez-Bello A., Uriarte E. et al. QSAR study for macromolecular RNA folded secondary structures of mycobacterial promoters with low sequence homology. (2005) 9th International Electronic Conference on Synthetic Organic Chemistry
[21] P. M. O'Grady, M. G. Kidwell. Phylogeny of the Subgenus Sophophora (Díptera: Drosophilidae) Based on Combined Analysis of Nuclear and Mitochondrial Sequences. (2001) Molecular Phylogenetics and Evolution Vol. 22, No. 3,
[22] Bao-cheng Wang , Jecheol Park , Hide-aki Watabe et al. . Molecular phylogeny of the Drosophila virilis section (Díptera: Drosophilidae) based on mitochondrial and nuclear sequences. (2006) Molecular Phylogenetics and Evolution vol. 40
[23] Wen-Ya Ko, Ryan M. David, Hiroshi Akashi .Molecular Phylogeny of the Drosophila melanogaster Species Subgroup. (2003) Journal of Molecular Evolution.
[24] P. S. Jefs,' E. C. Holmes, 2 and Al. Ashburner. The Molecular Evolution of the Alcohol Dehydrogenase and Alcohol Dehydrogenase-related Genes in the Drosophila melanogaster Species Subgroup. (1994) Molecular Biology Evolution
[25] K. Van Der Lindel, D. Houle , G. Spicer et Al. A supermatrix-based molecular phylogeny of the family Drosophilidae. (2010) Genet. Res., Camb.
[26] http://www.mirbase.org/cgi-bin [ Links ]

