










Services on Demand
Journal
Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO  uBio
uBio
Share

 Permalink
PermalinkRevista Boliviana de Química
On-line version ISSN 0250-5460
Rev. Bol. Quim vol.22 no.1 La Paz 2005
ARTÍCULO ORIGINAL
VALIDACIÓN DE UNA NUEVA DETERMINACIÓN ESPECTROFOTOMÉTRICA PARA DIPIRONA EN FÁRMACOS
Israel Quino a, Rigoberto Choque a, Luis Fernando Cáceres C b.
a Investigadores Asociados al CIN - IBTEN, Viacha, La Paz-Bolivia
b Centro de Investigaciones Nucleares, IBTEN, Viacha, La Paz-Bolivia
Dirección Autor Corresponsal: caceresluisfer@yahoo.es
RESUMEN
Se presenta un método para la determinación espectrofotométrica de la dipirona ([(1,5-dimetil-3-oxo-2-fenil-2,3 -dihidro- 1H-pirazol-4-il)-N-metilamino] metansulfonato) tanto sódica (DPNa) como magnésica (DPMg) por espectrofotometría de absorción molecular UV. El principio de determinación se basa en la absorción molecular de la dipirona en medio ácido a 257 nm. Ambos tipos de dipirona, presentan máximos de absorción entre 257-258 nm, con la única diferencia para la dipirona magnésica que presenta un otro pico de absorción a 293 nm. El método fue validado y cumple con todos los requerimientos de control y aseguramiento de calidad recomendados por la FDA (Food and Drugs Administration) de los EEUU. El método es selectivo y sensible, con límites de detección y cuantificación de 0,011 y 0,037 mg-L-1 para DPNa respectivamente y 0,036 y 0,120 mg-L-1 para la DPMg respectivamente, lo cual hace posible su cuantificación como impureza o traza en otros principios activos. Se obtienen respuestas lineales hasta 100 mg-L-1, con un r2 = 0,99919. Se verificó, la selectividad y robustez del método analítico, determinando el contenido de dipirona en tres formas farmacéuticas (comprimidos, gotas, e inyectables), provenientes de la industria farmacéutica tanto local como extranjera. En todos los casos, se encontraron porcentajes de recuperación de 92-111%.
ABSTRACT
We present a method for the spectrophotometric UV absorption determination of the dipirone ([(1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pirazol-4-il)-N-ethylammoniun] metansulfonate) as well as sodium (DPNa) and magnesium (DPMg) salts. The principle of determination is based on the molecular absorption of the dipirone in acid medium at 257 nm of wavelength. Both dipirone types, present a maximum of absorption between 257-258 nm, with the only difference for magnesic dipirone that presents another peak at 293 nm. The method was validated and it fulfills control requirements and insurance of quality recommended by the FDA (Food and Drugs Administration, USA). The method is selective and sensitive, with detection and quantification limits from 0,011 to 0,037 mgL-1 for DPNa, and 0,036 to 0,120 mgL-1 for the DPMg respectively, which makes possible quantification of for instance impurities or their traces of other active principles. Linear responses are obtained up to 100 mgL-1, with a r2 = 0.99919. The selectivity and robustness of the analytic method was verified, determining the dipirone content in three pharmaceutical forms (tablets, drops, and injectable), coming out from the local pharmaceutical industry as well as the foreign one. In all the cases, percentages of recovery were about 92 -111%.
INTRODUCCIÓN
La dipirona, (Figura 1) químicamente la [(1,5-dimetil-3-oxo-2-fenil-2,3-dihidro-1H-piridazol-4-il)-N-metilamino] metansulfonato, es empleada en formulaciones principalmente como analgésico. El método normal y recomendado para su determinación en fármacos, se basa en su reacción con una solución valorada de yodo 0,1 M [1], este método, no permite la cuantificación de cantidades a nivel de trazas.
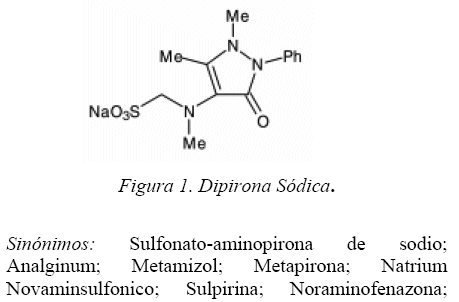
Sinónimos: Sulfonato-aminopirona de sodio; Analginum; Metamizol; Metapirona; Natrium Novaminsulfonico; Sulpirina; Noraminofenazona Metansulfonato Noramidopirona de sodio; Novamidazofen; Noramidazofenona [1].
En el presente estudio, tomo fármacos locales e importados, los cuales se describen en la Tabla 1, junto al principio activo y su forma farmacéutica.
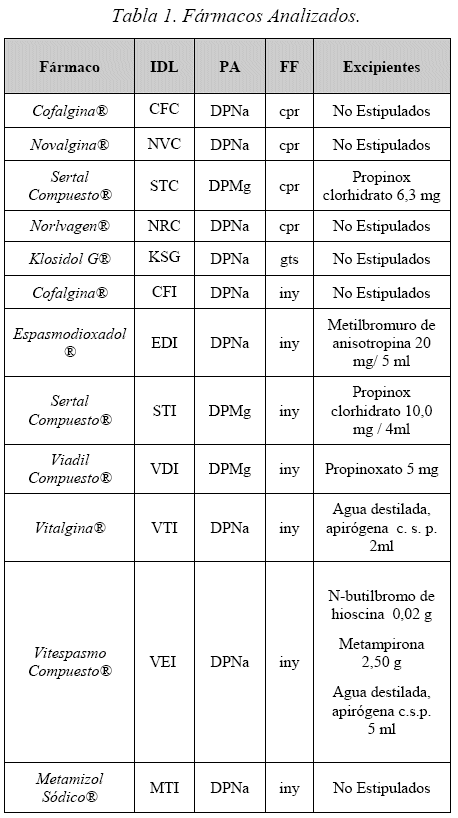
Las abreviaciones empleadas en la Tabla 1 son: PA: Principio Activo, FF: Forma Farmacéutica, DPNa: Dipirona Sódica, DPMg: Dipirona Magnésica, cpr: comprimido, gts: gotas, iny: inyectable. Todos los fármacos analizados, contienen 500 mg/Cpr en el caso de comprimidos y 500 mg/mL en el caso de gotas e inyectables; salvo el caso de Sertal Compuesto® comprimido que contiene 300 mg/cpr (Valores tomados de los folletos adjuntos a los productos).
EXPERIMENTAL
Reactivos y Soluciones
Todas las soluciones fueron preparadas usando agua destilada - desionizada con una resistividad de 18,2 Ωohm/cm, provistas por un sistema de purificación NANOpure® DiamondTM UV, se empleo HCl p.a. (37% m/v).
Las soluciones patrón de 1000 mg-L-1 de Dipirona Sódica y Magnésica se prepararon por disolución de Dipironas grado USP en HCl 0,1 M. Las soluciones de trabajo fueron preparadas diariamente por dilución de la solución patrón en HCl 0,1 M.
Los fármacos sometidos a estudio, fueron adquiridos en locales comerciales (farmacias) de la ciudad de La Paz.
Instrumentación
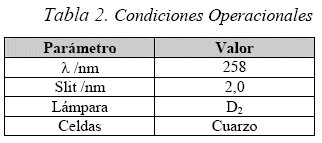
Para las determinaciones de absorción molecular se empleó un espectrofotómetro UV-VIS Helios-λ, doble haz de UNICAM. Todas las lecturas fueron realizadas con los parámetros de la Tabla 2.
Procedimientos
Los comprimidos fueron molidos en mortero de ágata hasta un tamaño de partícula menor a 0,3 mm, las soluciones fueron homogenizadas empleando un agitador magnético Lab-Line® Pyro-Magnestir.
Se tomaron alícuotas adecuadas de las soluciones tanto de inyectables como de gotas, empleando micropipetas automáticas eppendorf de 10-100 μl y 100-1000 μl de capacidad.
Validación del Método
El procedimiento fue validado contemplando los siguientes parámetros analíticos:
Linealidad
Definida como la capacidad para producir respuestas proporcionales a la concentración del analito [2].
Límite de Detección
Definido como la concentración de analito que produce una señal analítica igual al mínimo detectable, se calculó como:

Donde, b es la pendiente de la curva analítica y Sb la desviación estándar de la pendiente, calculada como:
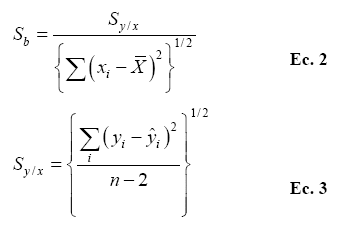
Donde Sy/x es la desviación estándar de la relación entre concentración xi y absorbancia yi [2].
Límite de Cuantificación
Definida como la cantidad más pequeña de analito en una muestra, que puede determinarse cuantitativamente con una precisión y exactitud adecuada [2].
Los valores de Sb y de b son calculados de igual forma que para el límite de detección.
Selectividad/Especificidad
La Selectividad es la capacidad de un método, bajo condiciones establecidas de ensayo para determinar exactamente el analito de interés en presencia de otros componentes en la matriz de la muestra [2]; mientras que la Especificidad es la capacidad de un método para medir solamente lo que se tiene que medir. Esto es, evaluar inequívocamente el analito en presencia de los componentes que se espera podrían estar presentes. Típicamente, esto puede incluir impurezas, degradantes, matriz, etc. Un método es específico, cuando presenta la máxima capacidad de selección [2].
Precisión
Es el grado de concordancia (proximidad) entre resultados de mediciones repetidas de la misma propiedad. La precisión de un método puede establecerse a partir de la repetibilidad y reproducibilidad de los resultados [2].
La repetibilidad de un método es la proximidad de concordancia entre los resultados de mediciones sucesivas del mismo mensurando realizadas bajo las mismas condiciones de medición [2].
Las condiciones de repetibilidad incluyen: El mismo procedimiento de medición, el mismo observador u operador, el mismo instrumento de medición utilizado bajo las mismas condiciones, el mismo lugar (mismo laboratorio), y repetición en un corto intervalo de tiempo.
La reproducibilidad puede ser expresada cuantitativamente, como la desviación estándar de los resultados obtenidos por distintos laboratorios en los resultados de un mismo ensayo, prueba o análisis [2].
Las condiciones que pueden variar incluyen: Principio y método de medición, operador, instrumento de medición, patrón de referencia, lugar (laboratorio), condiciones de uso y tiempo.
Exactitud
Proximidad entre el resultado de una medida o de la media de un conjunto de los mismos y el valor verdadero o valor convencionalmente verdadero del mensurando, expresado generalmente en función del error [2].
En el caso de un análisis rutinario, la exactitud será igual a la diferencia entre el valor medio obtenido al analizar un MRC (Material de Referencia Certificado) y su valor certificado (diferencia expresada como porcentaje del valor certificado).
Robustez
Medida de la capacidad de un procedimiento de medición química de permanecer inalterado por pequeñas, pero deliberadas variaciones en los parámetros del método y proveer una indicación de su confiabilidad durante un ensayo normal [2].
RESULTADOS
Linealidad
Se evaluaron 2 variantes, la primera construyendo curvas con los fármacos en el rango establecido y la segunda empleando los principios activos puros.
1ra Prueba. Curvas analíticas con los fármacos.
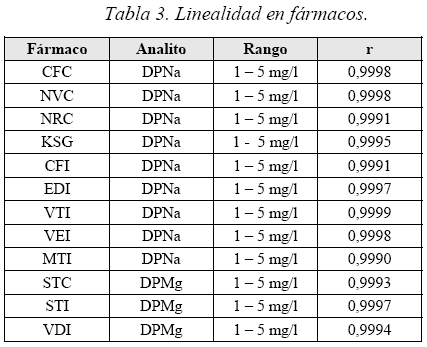
2da Prueba. Curvas analíticas con los principios activos. Se prepararon soluciones de 2,5; 4,0; 5,0 y 6,0 mg-L-1 (50%, 80%, 100% y 120% respectivamente del patrón de referencia), obteniéndose la respectiva curva de calibración.

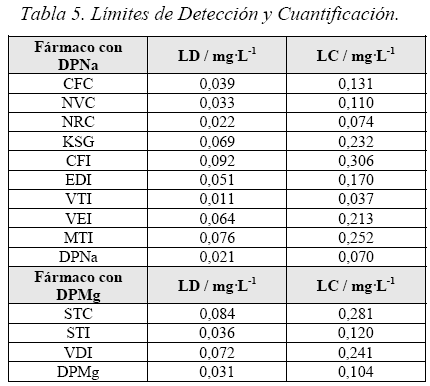
Límite de Detección y Límite de Cuantificación
Se prepararon soluciones de DPNa y DPMg de 1 - 5 mg-L-1, a partir de una solución inicial de 1000 mg-L-1. Luego se prepararon muestras de las mismas concentraciones de todos las muestras estudiadas, para hallar sus curvas de calibración en el espectrofotómetro a λ = 258 nm. A partir de los valores obtenidos en cada curva de calibración hallamos el Límite de Detección (LD) y el Límite de Cuantificación (LC), usando para el cálculo las ecuaciones 1 y 4.
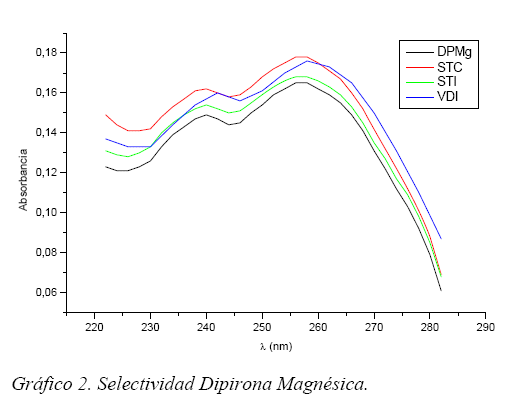
Selectividad/Especificidad
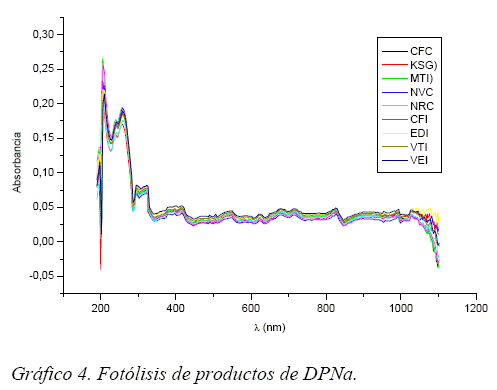
La selectividad del método esta demostrado por el espectro UV obtenido para cada muestra, mostrando el pico absorción del analito respectivo comparándolo con el pico absorción de los analitos puros. Observándose de esta forma las interferencias por los excipientes u otras impurezas. (Gráficos 1 y 2)
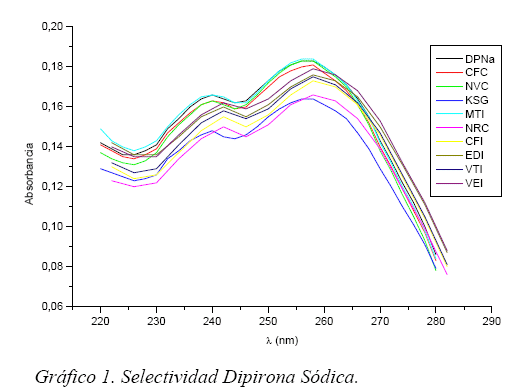
La selectividad de la DPNa es mayor para aquellos fármacos que tienen mayor recuperación, por ejemplo en el caso de NVC este presenta una mayor selectividad casi similar a la de la DPNa pura y su recuperación es del 109,7 % ocurre lo contrario para los fármacos que tienen menor recuperación como NRC que presenta menor selectividad y su recuperación es de 92,1%.
La selectividad de la DPMg esta relacionada también con la recuperación de esta en el método, como observamos la selectividad es mayor para STI ya que este tiene una mayor recuperación 99% en cambio la selectividad de los otros dos fármacos es menor al igual que su recuperación 96,4% de VDI y 93,1% de STC.
Precisión / Repetibilidad
Realizada por el mismo ensayista, en dos días diferentes. Se prepararon 6 soluciones de la misma concentración (5 mg-L-1), para todas las muestras y se hizo la lectura de estas soluciones por espectrofotometría observando la repetibilidad de las lecturas, luego comparamos los valores de absorbancia de las muestras con los valores de los analitos puros en función de su concentración para hallar la Desviación Estándar Relativa (%DER) de cada uno (Tabla 6).
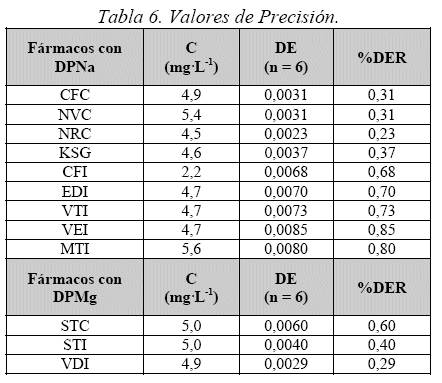
Como se puede observar, los resultados obtenidos, muestran en todos los casos un %DER por debajo del 1%.

Los resultados obtenidos para esta prueba presentan un %DER menor al 1,5.
Exactitud
Se prepararon patrones de 25 mg-L-1 de DPNa y DPMg. Las muestras se prepararon a la misma concentración pero se les adicionó DPNa y DPMg en un 50 % de su concentración (12,5 mg-L-1) obteniendo la recuperación de cada analito aplicando la ecuación 5 y descrita en la Tabla 8.
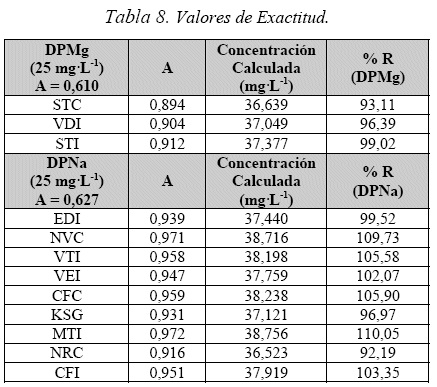
Para las muestras estudiadas el valor de recuperación esta dentro de 93,1 - 99,0% para DPMg y 92,2 -110,0% para DPNa.
Robustez
Se realizaron pruebas de Fotólisis, Termólisis y estabilidad en función del pH.
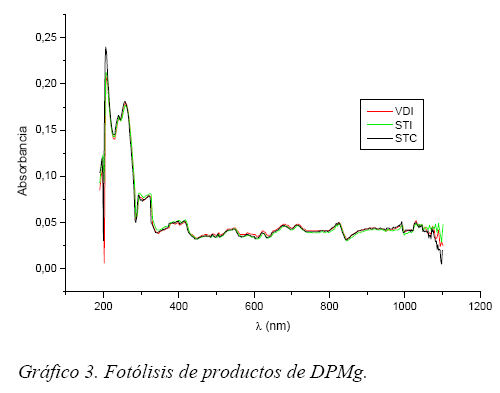
Fotólisis. Se irradiaron muestras de 5 mg-L-1 de concentración, variando el tiempo de exposición entre 0 a 60 minutos en intervalos de 10 minutos a temperatura constante. Esta prueba nos sirvió para observar la variación de los espectros de absorción de las diferentes muestras en función al tiempo de exposición a radiación ultravioleta de λ = 325 nm.
Los Gráficos 3 y 4 pertenecen a las muestras irradiadas durante 60 minutos, tanto los espectros para la DPMg como para DPNa son prácticamente similares, si bien en el caso de la DPNa existen ligeras variaciones atribuibles a la presencia de excipientes.
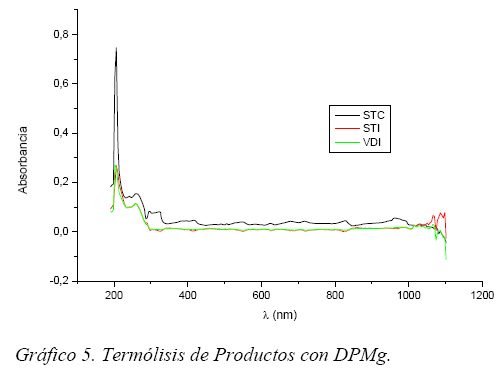
Termólisis. Se sometieron muestras de 4 mg-L-1 a variaciones de temperaturas desde 15 hasta 90°C en intervalos de 10°C, con plataformas de 10 minutos cada una.
En el Gráfico 5, se presenta el efecto térmico producido a 60° C sobre DPMg en los productos STI y VDI compuestos, ambos inyectables, al mismo tiempo, se aprecia que en STC comprimido los efectos sobre DPMg son menos acentuados que sobre los dos inyectables.
Como se aprecia en la Figura 6, el efecto térmico a 60°C sobre DPNa es mucho más acentuado, ya que los productos EDI, CFI, KSG, MTI, CFC, NRC y VEI, sufren alteraciones estructurales reflejadas en los espectros de absorción molecular, siendo que el NVC y VTI sufren un menor efecto.

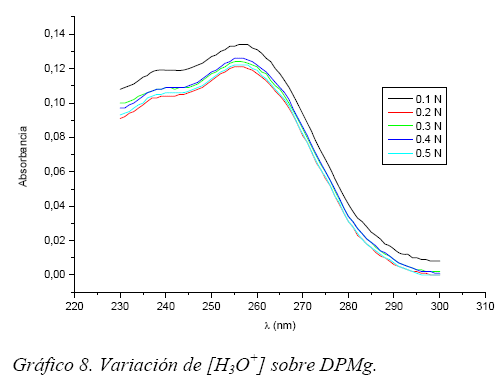
pH. Se prepararon 5 soluciones de 5 mg-L-1 cada una, tanto de DPMg como de DPNa, en medio de HCl concentraciones finales desde 0,1 hasta 0,5N.
Al variar la concentración de HCl (incremento de pH) en el caso de la DPNa y la Figura 7; la absorbancia crece hasta una concentración de 0,2 N y tiende a mantenerse constante para concentraciones mayores de ácido; ocurre lo contrario con la DPMg y la Figura 8, esto es, la absorbancia disminuye hasta una concentración de 0,2 N y tiende a mantenerse constante para concentraciones mayores.

CONCLUSIONES
El artículo que presentamos describe un nuevo método para la determinación de Dipirona Sódica y Magnésica en fármacos, mediante Espectrofotometría de Absorción Molecular. El método posee alta precisión, exactitud, rapidez, y sencillez, por lo cual se constituye en un método muy apropiado, ya que puede determinar de forma cuantitativa trazas o impurezas de Dipirona Sódica o Magnésica en diferentes formas farmacéuticas.
La linealidad del método es alta con un coeficiente de correlación de 0,999 como valor mínimo en las diferentes pruebas, el método es altamente selectivo con límites de detección de 0,011 y 0,036 mg-L-1 para DPNa y DPMg respectivamente y límites de cuantificación de 0,037 y 0,120 mg-L-1 para DPNa y DPMg respectivamente, lo cual nos muestra la factibilidad técnica del método.
La recuperación de los analitos en las muestras es alta con un rango de 90 - 110 % al igual que la precisión determinada como repetibilidad y reproducibilidad de resultados con un %DER menor al 1%.
La exposición a la radiación ultravioleta no altera la determinación de dipirona, la temperatura es un parámetro a tomar en cuenta porque al incrementarse ésta, algunas muestras sufren alteraciones en sus espectros de absorción, debido posiblemente a la composición propia de cada fármaco.
Se establece que la concentración mínima de HCl a emplearse en la determinación, es de 0,2N para ambos principios activos.
Estas tres últimas pruebas nos llevan a concluir el buen grado de robustez del método.
Finalmente como parte del trabajo se pretendió comparar contra un método volumétrico, lo cual no fue posible ya que las concentraciones de trabajo eran muy bajas y el método (yodometría) requiere de concentraciones mucho más altas.
AGRADECIMIENTOS
Los autores agradecen especialmente la valiosa y profesional colaboración de la Lic. Magaly Carmen Iriarte V.
Al LQA - CIN - IBTEN por su incentivo y apoyo a la investigación.
REFERENCIAS
[1] United States Pharmacopeia, USP-NF, XXIV, 2002. [ Links ]
[2] Cáceres C. L.F., Glosario de Términos en Análisis Químico Ambiental,Para Publicación. [ Links ]

