











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
La esclerosis lateral amiotrófica (ELA) es una enfermedad neurodegenerativa caracterizada por la muerte progresiva de neuronas motoras y que se asocia a un deterioro extramotor de intensidad variable. Su descripción inicial tuvo lugar gracias a Jean-Martin Charcot, el cual observó lesiones a nivel de los cordones laterales mismos que ocasionaban contracturas, lo que ahora se denomina síndrome piramidal, y que la afectación de las astas anteriores provocaba atrofia, su trabajo adquiere impacto al sintetizar estas observaciones y reconocer su asociación en una entidad nosológica desconocida hasta ese entonces (1). Posteriormente, junto a su discípulo Alix Joffroy quien más tarde sería reconocido como uno de los pioneros de la neuropatología, Charcot llevó a cabo en 1869 la descripción del síndrome que llevaría su nombre. En sus palabras: “Aquí, la lesión medular se caracteriza por la combinación de una alteración constante de la sustancia gris anterior con una esclerosis primaria y simétrica de los cordones laterales de la médula espinal.”(2).
No sería sino hasta 1874 cuando le dio a la enfermedad el nombre de esclerosis lateral amiotrófica, de la cual dejó una amplia descripción en las lecciones duodécima y decimotercera sobre las enfermedades del sistema nervioso, en el volumen II de las Obras completas publicadas en 1886 por Bourneville. En la actualidad la ELA sigue siendo una entidad de etiología desconocida, excepto en los casos en los que rara vez se puede determinar la mutación genética patógena, sin embargo, se ha establecido que en la gran mayoría de cuadros clínicos existe inclusiones citoplasmáticas en las neuronas afectadas a partir de una proteína llamada TDP43, la cual altera la homeostasis de los ácidos nucleicos, en este mismo contexto, la degeneración lobar frontotemporal muestra el mismo patrón patogénico, lo cual entraña una relación entre ambas patologías (3).
La incidencia y prevalencia de la ELA varía de acuerdo a cada región, por ejemplo, para Europa occidental, un metaanálisis evidenció una incidencia mediana de 2,08 y una prevalencia mediana de 5,40/100.000 habitantes (4). Cabe mencionar que, en el Pacífico Occidental existen brotes endémicos de ELA asociados a enfermedad de Parkinson y demencia, cuya etiología probablemente se deba a un aminoácido no proteico con propiedades excitotóxicas, la BMAA (beta-N-methylamino-L-alanine), la cual fue descubierta en los años 50 en la isla de Guam en las nueces de cícadas, su mecanismo patogénico involucra una activación de receptores del glutamato (NMDA y AMPA), provocando una entrada excesiva de calcio y muerte neuronal, el estrés oxidativo que se genera promueve la formación de radicales libres y daño celular, así también puede sustituir a la L-serina en la síntesis proteica, lo que daría lugar a proteínas mal plegadas que contribuyen a la neurodegeneración (5).
En la década de 1950, se documentaron varias cohortes distintas de esclerosis lateral amiotrófica (ELA), la más prominente de las cuales se observó en Guam durante un período en el que la incidencia de casos superó los niveles previstos en la población local. En la actualidad, ya no existe una mayor prevalencia de ELA en Guam, y la causa subyacente del aumento observado anteriormente, ya sea de origen genético o ambiental, sigue siendo un área de investigación en curso, y se han propuesto numerosas teorías a lo largo de los años (6). Es muy probable que los determinantes ambientales por sí solos sean insuficientes para precipitar la enfermedad; más bien, se cree que interactúan de forma sinérgica con la predisposición genética. Del mismo modo, la hipótesis de que la ELA puede estar asociada con una infección viral es una noción que resurge periódicamente en el discurso académico (7).
Más allá de la clásica afectación motora de predominio piramidal, la ELA puede manifestarse con fenotipos clínicos mixtos, donde los síntomas extrapiramidales como: la rigidez, bradicinesia o posturas distónicas desafían los criterios diagnósticos convencionales y pueden simular parkinsonismo primario o secundario. Este espectro clínico, conocido como ELA con fenotipo extrapiramidal no Guam, plantea un reto diagnóstico incluso para especialistas experimentados, dado que la coexistencia de signos de motoneurona superior e inferior con pobre respuesta a levodopa obliga a reconsiderar el diagnóstico diferencial frente a enfermedades neurodegenerativas dopaminérgicas, síndromes paraneoplásicos y procesos metabólicos. En este contexto, el reconocimiento precoz de estos fenotipos atípicos de ELA resulta fundamental no solo para establecer un diagnóstico certero, sino también para orientar la estrategia terapéutica, el pronóstico funcional y el acompañamiento multidisciplinario del paciente.
El presente caso ilustra una forma progresiva de ELA con rasgos extrapiramidales y hallazgos clínicos y electrofisiológicos distintivos, en una paciente sin exposición a neurotoxinas ni antecedentes familiares, subrayando la necesidad de ampliar la mirada más allá del paradigma clásico del Parkinson.
DESCRIPCIÓN DEL CASO CLÍNICO
Paciente femenina de 64 años de edad, casada, de religión católica, con instrucción primaria incompleta, residente en zona rural cercana a la ciudad de Cuenca, quien acude a hospital público el 28 de septiembre de 2025. Presenta antecedentes personales de hipertensión arterial sistémica en tratamiento con Losartán 100 mg VO QD, estreñimiento crónico y síntomas urinarios de larga evolución (polaquiuria, disuria leve intermitente). Destaca un diagnóstico de “Enfermedad de Parkinson” realizado por médico general tres meses atrás, con prescripción de levodopa/carbidopa que suspende por su cuenta tras pocas semanas de uso. No refiere alergias medicamentosas ni consumo de alcohol o tabaco. Acude por un cuadro progresivo de aproximadamente un año de evolución, caracterizado por parestesias intermitentes en miembros inferiores, astenia marcada, debilidad progresiva y deterioro de la marcha, que en las últimas semanas le impidió mantenerse en bipedestación. Asocia pérdida ponderal no cuantificada, estreñimiento persistente y sensación de inestabilidad postural.
Al ingreso, los exámenes iniciales evidenciaron hiponatremia severa (Na 120 mEq/L), con osmolaridad plasmática de 231.8 mOsm/kg y osmolaridad urinaria elevada, en una paciente clínicamente euvolémica, sin signos de deshidratación ni sobrecarga hídrica. La función renal, tiroidea y suprarrenal resultaron normales, también se descarta uso de diuréticos, insuficiencia renal, hepática y pérdidas digestivas. Frente a ello se planteó el diagnóstico de Síndrome de Secreción Inadecuada de Hormona Antidiurética (SIADH).
En Emergencias se inició manejo con solución salina hipertónica al 3%, logrando una mejoría parcial del sodio (hasta 126 mEq/L) en las primeras 48 horas; sin embargo, ante la falta de respuesta sostenida, se instauró restricción hídrica (500 ml/24 h) y suplementación con cápsulas de sal, obteniendo finalmente una corrección estable del sodio (134-135 mEq/L) y mejoría clínica del cuadro metabólico. Inicialmente recibió antibioticoterapia empírica con ceftriaxona por sospecha de infección urinaria, la cual se descartó al considerarla bacteriuria asintomática (urocultivo negativo, sin fiebre ni leucocitosis, sin síntomas urinarios irritativos actuales).
A la evaluación neurológica, la paciente se encontraba vigil, atenta, orientada en tiempo, espacio y persona, con memoria a corto y largo plazo conservada y funciones cognitivas superiores preservadas (considerando paciente de baja escolaridad). No se evidenció compromiso de pares craneales. Llamó la atención una facies inexpresiva con acentuación de comisuras labiales y voz espástica de tono alto, sin alteración en la fluidez ni en el contenido del lenguaje. En las extremidades se observó desviación cubital de ambas manos sin signos inflamatorios articulares, elevación del primer dedo de ambos pies (postura tipo estriatal), rigidez generalizada grado I con maniobra de distracción, acompañada de hipocinesia y bradipsiquia.
En las pruebas de coordinación y equilibrio, tanto el Romberg como el pull test resultaron positivos, con aumento de la base de sustentación, pasos cortos e inestabilidad postural, configurando una marcha espástica. Se objetivaron además fasciculaciones linguales discretas, posición distónica de manos y atrofia leve de eminencias tenares, sin alteraciones oculomotoras ni disfagia. La fuerza muscular global se mantenía conservada, con disminución leve proximal y distal en miembros inferiores, flexores y extensores (Daniels 4+/5). Apareció temblor de reposo, leve e intermitente en miembros superiores e inferiores, con marcha espástica y postura en camptocormia, además de dificultad para incorporarse desde la silla. En el examen de reflejos se evidenció hiperreflexia en el hemicuerpo izquierdo, junto con atrofia tenar e hipotenar y fasciculaciones linguales, hallazgos que combinan signos de motoneurona superior (espasticidad, hiperreflexia, rigidez) e inferior (atrofia, fasciculaciones), configurando un síndrome mixto piramidal-amiotrófico.
Durante la valoración pre y post administración de levodopa, se documentó rigidez leve generalizada y bradicinesia global leve a moderada (grados I-II), con una puntuación total MDS-UPDRS de 12 puntos en fase “off” y 10 puntos en fase “on”, lo que corresponde a una respuesta terapéutica negativa (<20%), descartando enfermedad de Parkinson. En conjunto, la ausencia de respuesta dopaminérgica, la progresión motora y la coexistencia de signos extrapiramidales y de motoneurona permitieron descartar parkinsonismo dopaminérgico y orientar el diagnóstico hacia Esclerosis Lateral Amiotrófica (ELA).
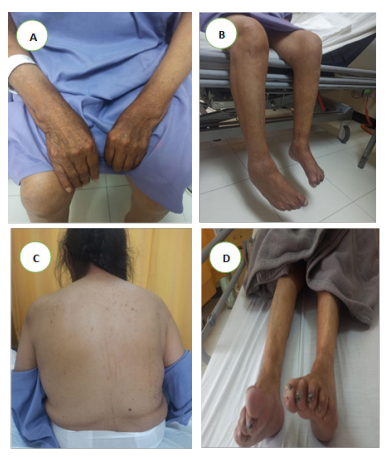
A. Se observa postura distónica con desviación cubital de ambas manos, sin signos inflamatorios articulares B. Se evidencia atrofia muscular distal bilateral, con predominio en los compartimentos anteriores y posteriores de las piernas, así como discreta rigidez y postura en extensión parcial de rodillas y flexión plantar leve. Estos hallazgos son coherentes con un patrón de debilidad distal progresiva C. En esta imagen se evidencia hipotrofia de masas musculares escapulares y paravertebrales, sin escoliosis ni deformidades estructurales evidentes. Esta atrofia axial, junto con la postura en ligera flexión de hombros, es consistente con debilidad proximal y pérdida del tono postural, contribuyendo a la camptocormia observada en la exploración neurológica. D. Imagen compatible con atrofia distal marcada con extensión del primer dedo y semiflexión de los restantes, configurando una posición tipo estriatal asociada a rigidez y aumento del tono muscular, sin lesiones tróficas ni edema.

La resonancia magnética cerebral evidenció atrofia córtico-subcortical leve y microangiopatía grado I de Fazekas, mientras que las resonancias cervical y lumbar mostraron discopatía degenerativa sin compresión radicular significativa, descartando causas estructurales que justificaran el cuadro motor. Dada la presencia de un síndrome constitucional con pérdida ponderal progresiva, se consideró la posibilidad de un síndrome paraneoplásico o enfermedad sistémica subyacente; por tal motivo, se realizó un TAC Body (tórax, abdomen y pelvis con contraste), el cual no evidenció lesiones neoplásicas activas, identificando únicamente esteatosis hepática leve, páncreas atrófico con discreta dilatación del conducto de Wirsung (8 mm) y un aneurisma infrarrenal de 32 mm, sin masas, adenopatías ni signos de enfermedad metastásica.
La electromiografía (06/10/2025) demostró fibrilaciones, fasciculaciones y ondas agudas positivas en territorios cervical y lumbosacro, con reclutamiento reducido y afectación asimétrica del hemicuerpo izquierdo, hallazgos altamente sugestivos de enfermedad de motoneurona, estos hallazgos sumados al test terapéutico con levodopa, cuyo resultado fue negativo, permitieron descartar enfermedad de Parkinson o parkinsonismo secundario y más bien consolidaron finalmente el diagnóstico de Esclerosis Lateral Amiotrófica (ELA).
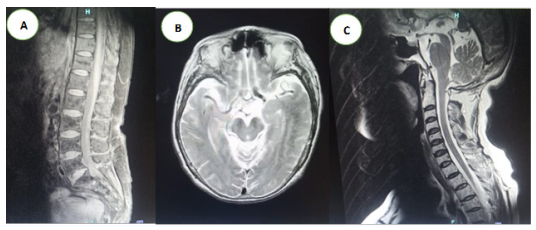
A. RMN columna lumbar. Signos de discopatía degenerativa en todos los segmentos lumbares. Protrusión multidireccional del anillo fibroso de L4-L5 y L5-S1 con compromiso moderado de raíces nerviosas bilaterales. La grasa epidural que recubre las emergencias nerviosas disminuida de espesor en L4-L5 y L5-S1 bilateral. Amplitud del canal raquídeo disminuida parcialmente en L4-L5 y L5-S1. Articulaciones interfacetarias esclerosas bilaterales. La médula espinal, el cono medular, el filum terminal y la cola de caballo se encuentran conservados.
B RMN cerebral corte axial. Hemisferios cerebrales con señal homogénea en todas las secuencias adecuada diferenciación entre sustancia gris y blanca. Surcos, cisuras de las convexidades y valles silvianos prominentes por involucion cortical a nivel de la sustancia blanca supratentorial subcortical bifrontal, biparietal, y periventricular se observa varios focos hiperintensos en secuencia T2 y Flair que no restringen la señal en secuencia de difusión DWI compatibles con proceso angiopático crónicos, adicionalmente se evidencia “Signo del Morning Glory”.
C. RMN cerebral corte sagital. “Signo del Colibrí Se instauró tratamiento con Riluzol 50 mg VO cada 12 horas y Biperideno 1 mg VO cada 12 horas, además de fisioterapia y fonoterapia. La paciente evolucionó lúcida, orientada y hemodinámicamente estable, con corrección definitiva del sodio (134-135 mEq/L), sin fiebre ni datos de infección activa. Fue dada de alta médica el 15 de octubre de 2025, con seguimiento ambulatorio por neurología, control de electrolitos séricos y medidas higiénico-dietéticas para el manejo del SIADH.
Discusión
La esclerosis lateral amiotrófica (ELA) se ha convertido en un ejemplo por excelencia de la neurodegeneración sistémica, en la que confluyen la pérdida de las neuronas motoras, el deterioro mitocondrial y la disminución de los circuitos neuronales corticales y subcorticales (8). La investigación fundamental de Neumann et al. (3) reveló que la ELA y la degeneración lobar frontotemporal muestran inclusiones citoplasmáticas compartidas que contienen TDP-43, recontextualizando así su fisiopatología dentro de un espectro de alteraciones relacionadas con el TDP-43 en el metabolismo del ARN y el desequilibrio proteostático.
En este marco, el paciente presentó un inicio clínico inusual, en el que predominaron las manifestaciones sistémicas: astenia, estreñimiento, debilidad muscular progresiva e hiponatremia pronunciada (Na 120 mEq/L), que se interpretó como un síndrome de secreción inadecuada de hormona antidiurética (SIADH). La presencia de hiponatremia euvolémica en el contexto de un trastorno neurológico progresivo planteó un dilema diagnóstico inicial, ya que podría indicar una disfunción hipotalámica, una infección sistémica o fenómenos paraneoplásicos (9). A esta complejidad se sumó la sospecha de infección urinaria, que posteriormente se excluyó, lo que retrasó la evaluación neurológica inicial. Este escenario subraya los desafíos inherentes a la identificación de la neurodegeneración multisistémica posterior a manifestaciones ostensiblemente metabólicas o infecciosas, algo que ocurre con frecuencia durante las etapas incipientes de la ELA extrapiramidal.
La evaluación neurológica subsiguiente reveló rigidez generalizada, bradicinesia, hipocinesia y posturas distónicas carentes de capacidad de respuesta dopaminérgica, acompañadas de fasciculaciones linguales y atrofia muscular distal, lo que refleja una presentación mixta con afectación de las neuronas motoras superiores e inferiores que delineó el síndrome piramidal-extrapiramidal. Este fenotipo, documentado en el 5 al 17% de los pacientes con ELA (10), se atribuye a disfunciones en los sistemas nigroestriatal y serotoninérgico como resultado de la pérdida neuronal tanto en los ganglios basales como en los núcleos del rafe (11). La resonancia magnética mostró indicadores indirectos de degeneración corticobasal, en particular el “signo del colibrí”, mientras que la electromiografía validó la denervación tanto activa como crónica, cumpliendo con los criterios establecidos en las versiones revisadas de El Escorial y Awaji-Shima (12,13).
Profundizando en los hallazgos de imagen la resonancia magnética cerebral mostró atrofia cortical difusa leve y signos morfológicos como el “signo del colibrí” y el “signo del Morning Glory”. El primero se describe clásicamente en la parálisis supranuclear progresiva (PSP), mientras que el segundo puede observarse en la atrofia multisistémica (MSA)(14). Sin embargo, ambos hallazgos pueden coexistir en ELA con afectación extrapiramidal, reflejando degeneración mesencefálica y del tegmento pontino sin hallazgos específicos de PSP o MSA. En este caso, su presencia se interpreta como un marcador de neurodegeneración multisistémica dentro del espectro ELA, más que como evidencia de otra entidad. Esta correspondencia entre clínica y neuroimagen apoya la hipótesis de que la ELA puede comprometer estructuras mesodiencefálicas y ganglios basales en fenotipos extrapiramidales(12).
A diferencia del complejo ELA-Parkinson y demencia observado en Guam, que está relacionado con la exposición al BMAA en las cícadas, según lo descrito por Garruto et al. (15) y Cox y Banack (16) este paciente carecía de antecedentes de exposición ambiental o predisposición familiar, lo que implica una etiología esporádica probablemente mediada por una disfunción mitocondrial. Las mutaciones en el POLG, según lo delineado por Stumpf et al. (17), se han asociado con fenotipos clínicos mixtos y déficits cognitivos leves, lo que refuerza la hipótesis de una perturbación subyacente en el metabolismo energético neuronal.
El tratamiento clínico de este paciente ejemplifica cómo una afección inicialmente percibida como metabólica o infecciosa puede revelar intrincados procesos neurodegenerativos. La necesidad imperiosa de sintetizar los datos metabólicos, electrofisiológicos y de diagnóstico por imágenes transformó el caso en un auténtico desafío diagnóstico multidimensional.
Desde una perspectiva terapéutica, el riluzol y el biperideno se instituyeron junto con las intervenciones de rehabilitación, estrategias que, si bien no alteran la trayectoria de la enfermedad, tienen como objetivo mantener la funcionalidad y mejorar la calidad de vida (18). De acuerdo con Matamala et al. (19), el manejo integral de la ELA debe basarse en un modelo multidisciplinario coordinado, donde intervienen neurología, fisioterapia, terapia ocupacional, fonoaudiología, nutrición y cuidados respiratorios. Este enfoque ha demostrado prolongar la sobrevida y reducir complicaciones frente a la atención convencional.
En fases avanzadas, la instauración oportuna de ventilación no invasiva, el manejo nutricional con gastrostomía cuando sea necesario y la participación temprana de cuidados paliativos son medidas que impactan directamente en la calidad de vida y en la toma de decisiones compartidas con el paciente y su familia.
En conclusión, este caso pone en evidencia que la ELA extrapiramidal no Guam es un reto clínico y diagnóstico progresivo, donde los límites entre metabolismo, infección y neurodegeneración se difuminan. Reconocer esta complejidad es esencial para replantear los enfoques diagnósticos y comprender que la ELA no es una enfermedad aislada de la motoneurona, sino una patología de red neuronal con expresión multisistémica y fenotipos en expansión.

