











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO  uBio
uBio 
 Permalink
Permalink
INTRODUCCIÓN
A finales del año 2019 en la ciudad de Wuhan, China, se originó la enfermedad COVID-19, que es provocada según Zhu, N. et. al (2019) por el nuevo coronavirus SARS-CoV-2. Este virus se extendió con rapidez por todo el mundo y en marzo del 2020 fue declarada como pandemia por la Organización Mundial de la Salud OMS (WHO, 2020). Hasta el momento no existe un tratamiento definitivo, sin embargo, han surgido muchas propuestas, entre ellas la cloroquina, hidroxicloroquina, azitromicina, ivermectina, corticoides, interferón beta 1a (IFN-β 1a), lopinavir, ritonavir, tociluzimab, remdesivir, favipavir, umifenovir, plasma convaleciente, por mencionar algunos, pero hasta el momento ninguno ha demostrado efectividad comprobada como menciona Cai Q., et. al (2020).
En la visión de Vellingiri (2020) las plantas tienen actividad antiviral positiva in vitro e in vivo, se ha observado que su efecto puede ser diferente contra los virus RNA o DNA, con o sin envoltura, incluso contra diferentes tipos o cepas de un virus.
El eucalipto es un árbol perenne que pertenece a la familia Myrtaceae, originaria de Australia, actualmente distribuida por todo el mundo. La hoja de eucalipto, a través de su compuesto principal el eucaliptol (1,8-cineol) ha demostrado su efecto antiviral en los estudios de Li, Y. et. al. (2016) sobre la influenza-A H1N1 a través de la inhibición del RNAm, Yang, Z. et. al. (2010) estudio la actividad del eucaliptol sobre el virus de la bronquitis infecciosa aviar, un tipo de coronavirus, mediante la inhibición del ingreso en fase de penetración y de replicación viral.
En marzo del 2020, Sharma, A y Kaur reportó un estudio in silico en el que se describe la posible capacidad del eucaliptol de actuar sobre la proteasa principal del coronavirus 3CLpro (Mpro/proteasa similar a la quimotripsina) proteasa importante en la replicación viral del SARS-CoV-2. En mayo del 2020 los mismos autores Sharma, A y Kaur publican un trabajo sobre las moléculas bioactivas del aceite esencial del eucalipto, como potenciales inhibidores del coronavirus. En los coronavirus la proteasa Mpro está implicada en la maduración proteolítica del virus y se ha examinado como posible diana mediante la inhibición de la escisión de la poliproteína viral y así prevenir la propagación del virus. Las secuencias de las proteínas Mpro del SARS-CoV y del SARS-CoV-2 son idénticas en un 96% de forma que es plausible que el eucaliptol pudiera actuar sobre el nuevo coronavirus. Mediante simulación computacional se pueden obtener diferentes grados de unión entre los receptores de la proteasa y el ligando, en este caso el eucaliptol. Los resultados que reportan Sharma y Kaur (2020) muestran una afinidad entre el eucaliptol y la estructura proteica.
El acoplamiento molecular o docking molecular es una aplicación de la mecánica molecular utilizada para predecir energías y modos de enlace entre ligandos y proteínas, proporciona información de gran utilidad para el estudio de nuevos compuestos con efectos terapéuticos; en estudios donde se aplica el acoplamiento molecular, usualmente la mejor conformación es considerada como aquella que reporta la menor energía de unión, alternativamente puede ser seleccionada del cluster más poblado entre los clusters obtenidos después del acoplamiento molecular y también considerando el menor valor de RMSD (Root Mean Square Deviation) con respecto a una estructura de referencia. Según Latorraca, N. et al (2016) cuando se trabaja con proteínas y particularmente con enzimas como moléculas blanco, deben considerarse tres factores importantes: la especificidad o selectividad molecular de la proteína o enzima, la afinidad o fuerza con que se fija el sustrato o ligando a ella y la geometría del sitio de unión. El presente trabajo de investigación tiene el objetivo de encontrar la forma en la cual el 1,8-cineol se acopla a la proteasa viral Mpro/3CLpro del SARS-CoV-2.
MATERIAL Y MÉTODOS
En el acoplamiento molecular o docking molecular el algoritmo del programa o software computacional y/o servidor en línea toma al ligando que es la molécula en estudio generalmente pequeña y la coloca dentro del sitio activo de la proteína o blanco y se produce la unión o acoplamiento generando varias conformaciones (Sánchez, A., 2000). En este estudio se empleó la estructura del ligando eucaliptol ya modelizada en el trabajo de Nogales, J. & Choque, R, del 2023.
El blanco también llamado receptor o target constituye la proteasa viral Mpro/3CLpro del SARS-CoV-2 fue tomado de la base de datos del PDB (Protein Data Bank) con la designación 6LU7. Las moléculas de proteínas están indexadas en este banco de datos en forma purificada, cuyas estructuras están al estado cristalino y están asociados a moléculas de agua y algunas moléculas con las cuales han sido aisladas y depositadas en el PDB. Se procedió a la preparación del receptor o blanco, esta preparación del receptor consiste en la eliminación de moléculas de agua, cofactores y algunas moléculas con las que viene resuelta la estructura cristalina, disponiendo así de la estructura del blanco para el acoplamiento molecular.
Para la búsqueda de las propiedades geométricas y topológicas de la proteína, cavidades superficiales, internas y canales que incluyen al sitio activo en el blanco, se usó el servidor CASTp diseñado por Tian, W. et. al (2018) que ofrece servicios en línea y es ampliamente utilizado. Para el acoplamiento molecular se tomaron los siguientes parámetros: libre rotación de todos los enlaces del ligando con libertad conformacional, adición de cargas de Gasteiger a la proteína y al ligando, adición de hidrógenos a la proteína, generación de 100 poses para el ligando y utilización del Algoritmo Genético Lamarckiano (LGA). Este último se utilizó bajo las siguientes condiciones: 100 corridas del GA, 150 tamaño de población, 250000 máximo número de evaluaciones de energía.
Los programas empleados y servidores en línea fueron los siguientes: Software PyRx (https://sourceforge.net/projects/pyrx/) que utiliza los archivos de Autodock vina y autodock tools - Virtual Screening Tool, con el command prompt y con servidores en línea: 1-click docking (https://mcule.com/apps/1-click-docking/>), Covid 19 docking server (https://ncov.schanglab.org.cn), CB dock (https://cadd.labshare.cn/cb-dock2/php/blinddock.php), Docking server (https://www.dockingserver.com/web), Webina (https://durrantlab.pitt.edu/webina/), Amdock ( https://bio.tools/amdock) Assisted Molecular Docking with Autodock4 and Autodock Vina SOFT y el Command prompt o CMD que también utiliza los motores de Autodock vina y autodock tools; cada servicio requiere de los archivos de la proteína que actúa como blanco o diana en formato pdb y del archivo del ligando en este caso el eucaliptol.
Con cada software y o servidor en línea de acoplamiento molecular se realizaron 25 acoplamientos, haciendo un total de 200 acoplamientos moleculares.
Las visualizaciones se realizaron con los software Chimera 1.16 (https://www.cgl.ucsf.edu/chimera/); Discovery Studio 2021 (https://discover.3ds.com/discovery-studio-visualizer-download>); Molegro Molecular Viewer v7.0 2019 (http://molexus.io/molegro-molecular-viewer/), Schrodinger maestro 13.2 v2022 (https://www.schrodinger.com/products/maestro/); y Pymol 2.5.0 (https://pymol.org/2/)
RESULTADOS
Los análisis de docking muestran como resultado valores de Binding Affinity (BA) cuyas unidades son Kcal/mol, estos datos corresponden a la energía libre de Gibbs, mientras más bajo sea el valor, más alta será la afinidad entre el ligando y el receptor (Lee, M. et. al., 2006). En la Tabla 1, se muestran los valores de: la energía de unión o afinidad (BA), de RMSD (Root mean Square deviation o raíz de la desviación estándar cuadrada), de Kd (constante de disociación del docking), la eficiencia de enlace (E.L) y los aminoácidos involucrados en el acoplamiento entre el eucaliptol y la proteasa principal del SARS-Co-2 con los ocho servidores utilizados para el acoplamiento.
Con las conformaciones obtenidas se obtuvo la energía libre de Gibbs para la unión entre el eucaliptol y la proteasa viral, parámetro energético que es calculado por los programas respectivos y presentada en forma de afinidad energética BA.
En el presente docking molecular, se obtuvieron las conformaciones del eucaliptol en el sitio activo de la proteasa del SARS-CoV-2, seis de las conformaciones obtenidas con seis servidores y/o software presentan una energía de unión con valores de -4,5 Kcal/mol (ver tabla1) valor de acoplamiento que bajo los criterios establecidos por Lee, M y Olson, (2006) afirman que mientras más bajo sea el valor, más alta será la afinidad entre el ligando y el receptor.
Podríamos afirmar que esta conformación obtenida del eucaliptol es la más probable, sin embargo, hasta la fecha (10-10-2024) en el banco de datos de proteínas (PDB) no está reportado la molécula de eucaliptol acoplada con la proteasa principal del SARS-CoV-2, por tal motivo no se pudo realizar una comparación con un modelo experimental.
Al respecto Kar Bipasa et. al. en 2022 estudió trescientos fitocompuestosin silico y seleccionó tres para un análisis detallado de la interacción molecular con la Mpro del SARS-Cov-2. Estos fitocompuestos fueron la oleanderolida, proceragenina A y balsaminona A, los cuales presentaron energías de unión BA de acoplamiento de -9,4 kcal/mol, -8,6 kcal/mol y -8,1 kcal/mol respectivamente contra la proteasa principal en el mismo sitio activo.
Así mismo, en la tabla 1 se muestran los valores de RMSD, destacamos que los valores en cada servidor y/o programa e interfaz dieron el valor de cero, fueron esas estructuras conformacionales seleccionadas, al no contar con la estructura cristalográfica de acoplamiento entre el eucaliptol y la Mpro del SARS-CoV-2 en el banco de datos de proteínas, se procedió a recalcular nuevamente los valores de RMSD comparando con la estructura química conformacional obtenida con el prompt CMD.
Tabla 1 Energía de unión, RMSD, constante de disociación del docking, eficiencia de enlace (L.E) entre el eucaliptol y la Mpro del SARS-CoV-2
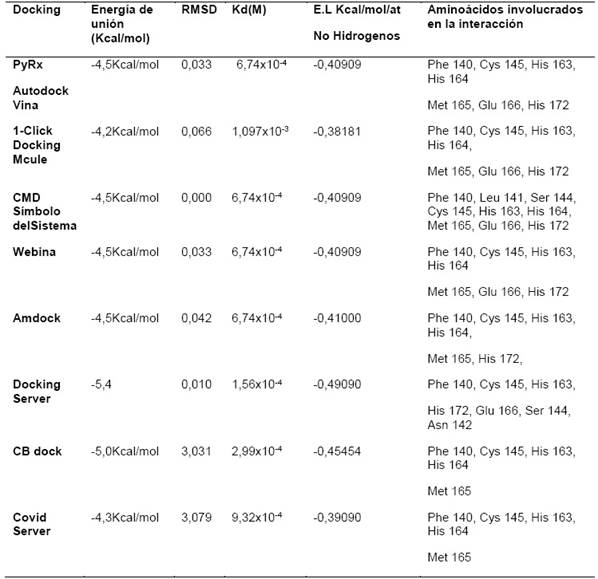
Energía de unión, RMSD, constante de disociación, eficiencia de enlace (E.L) del docking entre el eucaliptol y la Mpro del SARS-CoV-2 encontrados con los 8 programas de docking molecular. Los RMSD se calcularon en base al CMD
Mediante el uso de la expresión termodinámica: ΔGº = - RT Ln Kd, se calculó la constante de equilibrio Kd para cada una de las interacciones. Como se puede apreciar las constantes Kd tienen valores expresados en molar muy bajos, comportamiento que destaca Christopher W. Murray (2014) en sus trabajos de validaciones de ligandos, basándonos en estos hallazgos podemos afirmar que la molécula de eucaliptol y su objetivo biológico el sitio activo de la Mpro del SARS-CoV-2 se uniría fuertemente, este indicador de afinidad también se muestra en la tabla 1.
Otro enfoque es analizar el acoplamiento con el valor de la eficiencia de enlace E.L que representa una medida que relaciona la energía libre de unión con el tamaño de la molécula. Con la ecuación utilizada por Reynolds, C. (2011) E.L. = ΔGº / n donde E.L. representa a la eficiencia de enlace en Kcal/mol y n representa el número de átomos que no incluye a los átomos de hidrogeno. Los valores para este indicador de afinidad también se muestran en la tabla 1. Según la ecuación, a medida que aumenta la eficiencia de enlace, disminuye el peso molecular de la molécula en estudio, en nuestro caso el eucaliptol con un peso molecular de 154 g/mol constituye un ligando pequeño hecho que promovería una buena eficiencia de enlace. La E.L. es uno de los índices más utilizados en la actualidad, expresado en Kcal/mol/átomos que no son hidrogeno, proporciona una idea de la magnitud de energía de enlace por átomo distinto al de hidrogeno. Hay que tener presente que el valor de la eficiencia de enlace debe ser negativo, ya que la energía libre de unión es negativa. (Reynolds, C. y Holloway, K., 2011).
Las interacciones de carácter hidrofóbico detectados entre el eucaliptol y la proteasa viral se muestran en la tabla 2, en la que se aprecian los residuos de aminoácidos de la proteasa y las distancias entre el ligante eucaliptol y la proteasa viral que se presentaron con mayor frecuencia y en común con los ocho software y servidores en línea. Los aminoácidos en común con seis software y servidores en línea son la Phe y Glu excepto con CB dock y Covid Server donde se detectó la Asn en lugar de Phe. En un estudio sobre compuestos bioactivos realizado por Sharma, A. y Kaur, I. (2020) también detectaron al Glu y la Phe además de otros aminoácidos.
Tabla 2 Interacciones Hidrofóbicas entre el eucaliptol y la Mpro SARS-CoV-2
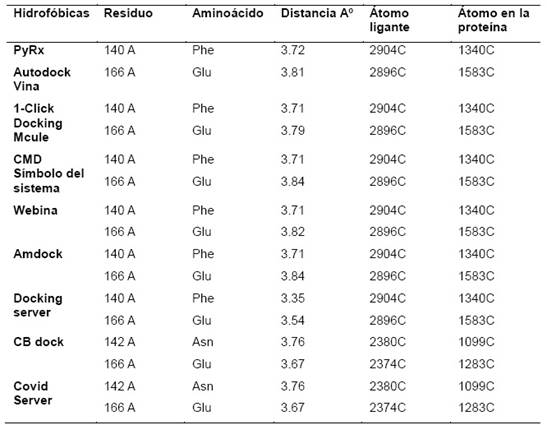
Interacciones detectadas con PLIP Protein Ligand Interaction Profiler https://plip-tool.biotec.tu-dresden.de/plip-web/plip/index) entre el eucaliptol y la Mpro del SARS-CoV-2 con los 8 softwares y servidores de acoplamiento.
En la Tabla 3 se presentan las interacciones detectadas por puente de hidrogeno, se indican los átomos donor y aceptor, esta interacción se dio entre el átomo de hidrogeno del N3 de la His y el único oxigeno del eucaliptol, esto se presentó en cinco softwares y/o servidores en línea, los 3 restantes servidores no presentaron el mencionado puente de hidrogeno, en la búsqueda bibliográfica no se encontraron trabajos in silico que puedan corroborar la interacción por puente de hidrogeno.
Tabla 3 Interacciones por puentes de hidrogeno detectadas con PLIP
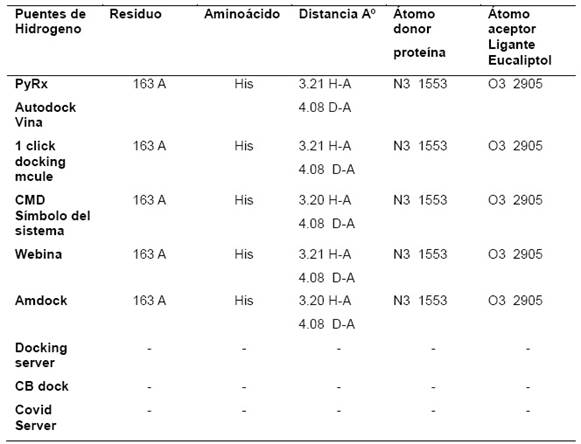
Interacciones detectadas con PLIP Protein Ligand Interaction Profiler https://plip-tool.biotec.tu-dresden.de/plip-web/plip/index) entre el eucaliptol y la main proteasa del SARS-CoV-2 con los 8 softwares y servidores de acoplamiento. PLIP
Las figuras 1 y 2 muestran el acoplamiento con PyRx, CMD, 1-Click docking, Amdock, Docking server, Webina, entre la Mpro del SARS-CoV-2 (en amarillo) y el eucaliptol (en magenta) visualizada en Chimera 1.16. Esta conformación se obtuvo con seis de los ocho servidores y programas utilizados para obtener el acoplamiento molecular, el eucaliptol está ubicado en el sitio activo más probable detectado en la proteasa viral.
Imagen obtenida en el docking molecular con 6 servidores (PyRx, CMD, 1-Click docking, Amdock, Docking server, Webina) entre la Mpro del SARS-CoV-2 (en amarillo) y el eucaliptol (en magenta). Visualizador Chimera 1.16. Ctrl+Click para ver video
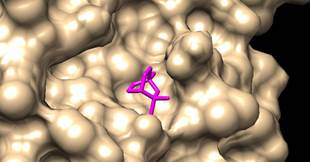
Acercamiento para visualizar el acoplamiento molecular con 6 servidores entre la Mpro del SARS-CoV-2 (en amarillo) y el eucaliptol (en magenta). Visualizador Chimera 1.16
Este sitio activo geométrica y topológicamente activo de la proteína, presento una cavidad superficial que incluye al sitio activo en este blanco, en la cadena A de la proteasa del SARS-CoV-2 el sitio activo presento un volumen de 226 Å3 con las coordenadas (x=-13.5, y=11.3, z=71.6) y los siguientes aminoácidos activos: Arg 188, His 41, Pro 168, Met 49, Cys 145, Thr 190, Asp 187, Glu 166, His 164, Tyr 54, Gln 189, Met 165, Leu 167, Gln 192; este sitio activo se determinó con el servidor CASTp que calcula la superficie topológica y geométrica en las proteínas. Kar Bipasa et. al. (2022), en estudios de acoplamiento de la oleanderolida, proceragenina A y balsaminona A detectaron el acoplamiento con la Mpro del SARS-CoV-2 en las coordenadas x=-13.1, y=15.87, z=70.36 muy similares a las que se utilizó en el presente estudio. Ramos, A. en 2021 confirmo los residuos del sitio activo de Mpro mediante un análisis estructural desarrollado con el software Wincoot, este análisis permitió establecer que los residuos que participan en la mayoría de las interacciones son: His41, Met49, Phe140, Leu141, Asn142, Gly143, Ser144, Cys145, His163, His164, Met165, Glu166, Asp187, Arg188 y Gln189, tomando como referencia la numeración de la secuencia de Mpro del SARS-CoV-2, lo cual muestra que son los aminoácidos encontrados en el presente estudio.
El acoplamiento de la Mpro del SARS CoV-2 (amarllo) y el eucaliptol (plomo) con el servidor Covid 19 server (figura 3a) y el eucaliptol (azul) con el servidor CBDock (figura 3b). Estos dos servidores dan resultados muy similares, encontrándose un RMSD entre ellos de 0,488, al no contar con el acoplamiento experimental en el banco de datos de proteína PDB, se compararon las estructuras obtenidas entre la estructura obtenida por CMD, el RMSD de estas dos estructuras puede apreciarse en la tabla 1, teniendo un valor de 3,031 y 3,079 valores altos para ser considerados como conformaciones ideales pero que tampoco podemos descartar ya que no podemos realizar la comparación con la estructura cristalizada del eucaliptol con la proteasa ya que esta no se encuentra en el PDB data bank.
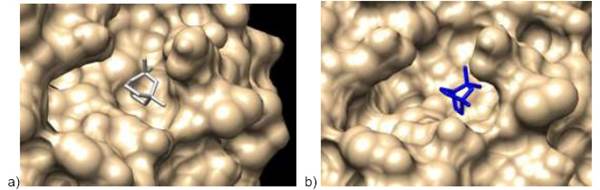
Imagen de acercamiento para visualizar el acoplamiento molecular entre Mpro del SARS CoV-2 (en amarillo) y: a) el eucaliptol (plomo) con el servidor Covid 19 server y en b) el eucaliptol (azul) con el servidor CBDock. Visualizador Chimera 1.16
El PLIP (Protein Ligand Interaction Profiler) (https://plip-tool.biotec.tu-dresden.de/plip-web/plip/index), es un perfilador de la interacción proteína ligando, utilizando esta herramienta se detectaron las interacciones por puente de hidrogeno e hidrofóbicas con diferentes softwares.
PLIP con PyRx, 1-click docking, CMD, Webina, Amdock y Docking server (figura 4) presentaron dos interacciones hidrofóbicas entre el ligando eucaliptol con los aminoácidos Phe 140 y Glu 166 de la Mpro; y un puente de hidrogeno entre el oxígeno del eucaliptol y el hidrogeno perteneciente al nitrógeno de la His 163; puente no detectado con CB dock y Covid server.
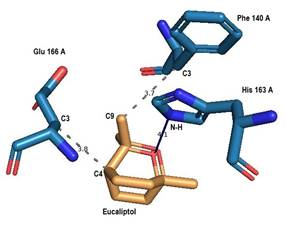
Figura 4 Interacciones por puente de Hidrogeno e hidrofóbicas detectadas con PLIP con PyRx, CMD, 1-Click docking, Amdock, Docking server y Webina
Interacciones detectadas con PLIP Protein Ligand Interaction Profiler https://plip-tool.biotec.tu-dresden.de/plip-web/plip/index) entre el eucaliptol y la Mpro del SARS-CoV-2 con 6 softwares y servidores de acoplamiento: PyRx, CMD, 1-Click docking, Amdock, Docking server y Webina. Líneas punteadas interacciones hidrofóbicas, línea continua puente de Hidrogeno.
PLIP con CB dock y Covid server (figura 5), en cambio mostraron dos interacciones hidrofóbicas entre el eucaliptol y los aminoácidos Asn 142 y Glu 166 de la Mpro del SARC-CoV-2.
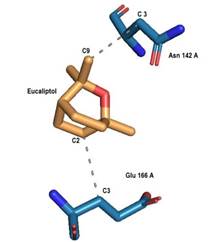
Interacciones detectadas con PLIP Protein Ligand Interaction Profiler https://plip-tool.biotec.tu-dresden.de/plip-web/plip/index) entre el eucaliptol y la main proteasa del SARS-CoV-2 con 2 servidores de acoplamiento, CBDock y Covid 19 server. Lineas punteadas interacciones hidrofóbicas.
En el estudio de Nogales J., y Choque R. (2023) se generaron propiedades moleculares y descriptores químicos que explicarían las interacciones entre el ligando eucaliptol y la proteasa principal del SARS-CoV-2 especialmente las fuerzas de atracción de Van Der Waals y las interacciones hidrofóbicas que es pertinente mencionarlos; el potencial químico µ para el eucaliptol describió que la densidad electrónica del sistema puede fluctuar, el flujo de electrones se dio de una zona de alto potencial químico a una región de menor potencial químico, el valor negativo encontrado µ=-2,19 eV mostro la variación espontanea de la densidad electrónica del sistema. Un descriptor muy relacionado al potencial químico de manera directa es la electronegatividad χ que para esta molécula mostro el mismo valor de signo contrario χ = 2,19eV indicando como la molécula tiende a atraer electrones sin cambiar su densidad electrónica; respecto a los orbitales frontera HOMO, HOMO-1, LUMO y LUMO+1 y los respectivos band-gap del eucaliptol, la densidad electrónica de los orbitales HOMO se concentra en la región del oxígeno del eucaliptol y los carbonos C6 C3 C4 C8 C9. El orbital LUMO en el eucaliptol presento una densidad electrónica distribuida sobre los carbonos del anillo C1 C2 C3 C4 C5 C6 mostrando orbitales deficitarios o zonas positivas en las regiones de los hidrógenos especialmente los anulares. La diferencia de energías de los orbitales frontera HOMO LUMO, se denomina band-gap, el valor hallado para el eucaliptol fue de 8,1 eV y la diferencia entre HOMO-1 y LUMO+1 dio 9,8 eV este valor sugiere que se necesita mucha energía para que ocurra una transición electrónica. Muy relacionado a estas diferencias, se encuentra la dureza que expresa la separación entre HOMO y LUMO, el valor de 4,05 eV para el eucaliptol sugiere una buena estabilidad del sistema químico con baja tendencia a dar o recibir electrones (Nogales, J. & Choque, R., 2023).
La interacción de un compuesto con una macromolécula depende en gran parte de sus características hidrofóbicas del ligando y de la superficie macromolecular, en el desarrollo del presente estudio se encontró que la interacción hidrofóbica es predominante, ya que la mayoría de los aminoácidos encontrados en este sitio activo son de carácter hidrofóbico. Los momentos hidrofóbicos según Eissenberg D. et al (1982) son igual a la suma vectorial de los momentos hidrofóbicos individuales de cada componente en una macromolécula, en nuestro caso los aminoácidos que forman parte del sitio activo de la proteasa del SARS-CoV-2. Una interacción como la hidrofóbica en la cual aparentemente no existen enlaces parecería que siempre es inespecífica, lo que significa que dos o más átomos cualesquiera con cierto carácter hidrofóbico interactuaran entre sí.
CONCLUSIÓNES
Los resultados de las simulaciones computacionales sugieren que el eucaliptol se une al sitio activo de la Mpro con una afinidad favorable, lo que podría tener implicaciones en el desarrollo de terapias antivirales. Existe una predominancia de fuerzas de interacción hidrofóbica intermolecular en el acoplamiento, ya que las características hidrofóbicas del sitio activo se deben a que la mayoría de los aminoácidos encontrados en este sitio activo son de carácter hidrofóbico
Es importante destacar que este estudio es preliminar y se basa en modelos computacionales. Se requiere validación experimental para confirmar la interacción entre el eucaliptol y la Mpro y determinar su potencial como agente antiviral contra el SARS-CoV-2. Con este estudio se abren nuevas líneas de investigación y destacamos la importancia de los estudios in silico y la generación de acoplamientos o interacciones entre dos moléculas. El eucaliptol, un compuesto natural con propiedades antivirales conocidas, podría ser una molécula candidata como fármaco preventivo y prometedor para el desarrollo de terapias contra el SARS-CoV-2, es necesario más estudios para confirmar su eficacia y seguridad.

