










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Científica Ciencia Médica
versión impresa ISSN 1817-7433versión On-line ISSN 2220-2234
Rev Cient Cienc Méd v.14 n.1 Cochabamba 2011
CASO
Síndrome de Hunter Mucopolisacaridosis (II): reporte de un caso
Hunter Syndrome Mucopolysaccharides (II): a Case Report
Arias Eulate Juan Carlos1, Angulo Flores Marcela Denisse1, Rueda Muñoz Zulma1, Ghunter Paz2
1Estudiantes de Medicina, Universidad Mayor de San Simón. Cochabamba, Bolivia.
2Docente de la cátedra de Neurología, Facultad de Medicina, Universidad Mayor de San Simón. Médico Neurólogo Pediatra, Hospital Obrero N°2 Caja Nacional de Salud, Cochabamba, Bolivia.
Correspondencia a: Juan Carlos Arias Eulate
carlitos_at77@hotmail.com
Procedencia y arbitraje: no comisionado, sometido a arbitraje externo.
Recibido para publicación: 23 de julio de 2011
Aceptado para publicación: 26 de septiembre de 2011
Citar como:
Rev Cient Cienc Med 2011;14(1): 40-42
RESUMEN
El síndrome de Hunter, es una alteración genética que afecta principalmente a los varones, debido a la deficiencia o ausencia de la enzima iduronato-2-sulfatasa, que interfiere con la capacidad del cuerpo de descomponer y reciclar los mucopolisacáridos. La incidencia es de 1: 10.000 a 1:25.000 de recién nacidos vivos. Las manifestaciones físicas, incluyen rasgos faciales distintivos, cabeza grande, abdomen aumentado, engrosamiento de válvulas cardíacas, enfermedad respiratoria obstructiva, retraso del desarrollo mental y aumento de tamaño del hígado y del bazo.
Presentamos el caso clínico de un paciente de sexo masculino de 7 años de edad, con diagnostico de síndrome de Hunter hace seis años, con antecedentes de crisis convulsivas en dos oportunidades y cuadros de bronconeumonía. Al examen físico presenta fascie tosca, contractura en musculo bíceps braquial, se logra la extensión de las manos, camina con la punta de los pies y presenta hepato y esplenomegalia. Al cual se le trató la sintomatología respiratoria con el uso de antibióticos.
Palabras claves: Síndrome de Hunter; Mucopolisacaridosis II; Iduronato Sulfatasa
ABSTRACT
Hunter Syndrome, is a genetic disorder that primarily affects males, due to the deficiency or absence of the enzyme iduronate-2-sulfatase, which interferes with the ability of the body break down and recyele mucopolysaccharides. The incidence is 1: 10.000 to 1:25.000 babies alive. The physical manifestations includes distinctive facial features, large head, abdomen increased thickening of heart valves, obstructive respiratory illness, delayed mental development and enlarged liver and spleen.
We present the clinical case of a 7-year-old male patient with diagnosis of Hunter Syndrome six years ago, with a history of seizures in two occasions and schedules of bronchopneumonia. A physical examination presents rough fascie, brachial bicep muscle contracture, is achieved by the extension of the hands, walks with the tip of toes and presents hepato and splenomegaly. Which we treated respiratory symptoms with the use of antibiotics.
Keywords: Hunter Syndrome; Mucopolysaccharidosis II; Iduronate Sulfatase
INTRODUCCIÓN
Las mucopolisacaridosis (MPS) y oligosacáridosis son un grupo de alteraciones metabólicas hereditarias debidas a una deficiencia de enzimas lisosómicas específicas, siendo la incidencia de 1:25 mil recién nacidos vivos1.
Las mucopolisacaridosis (MPS), se caracterizan por acumulación progresiva de glucosaminoglucanos (carbohidratos complejos que contienen amino azucares y ácidosuránicos). Estas moléculas se acumulan en los lisosomas de las células del tejido conectivo, incluido cartílago y hueso por deficiencia de enzimas lisosomales.
Las enzimas lisosomales rompen las largas cadenas de polisacáridos en unidades menores, dentro del lisosoma, las enzimas que intervienen en este proceso son diez y su deficiencia produce depósito intralisosomal de glucosaminoglucanos alterando la fisiología celular2.
La clasificación actual de la mucoplolisacaridosis comprende siete tipos 3 y es de acuerdo a la ausencia de la enzima. En el síndrome de hunter se encuentra afectada la enzima Iduronato -2 - sulfatasa (I2S) (tabla 1).
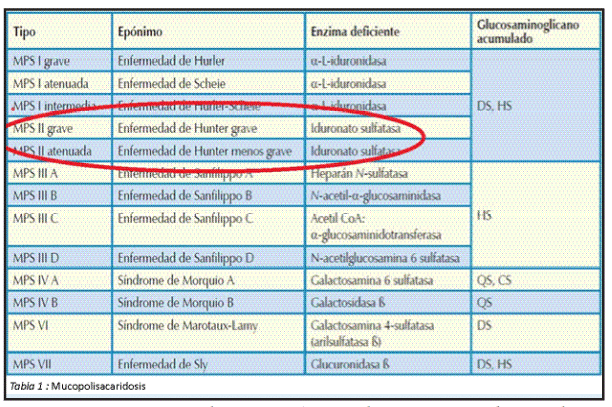
Se estima que el síndrome de hunter (MPS II) afecta a 1 en cada 155 mil nacidos vivos. El síndrome de hunter es un disturbio hereditario, recesivo, ligado al cromosoma x, que afecta principalmente personas del sexo masculino es por eso que es transmitida de una generación a otra de una manera especifica3.
En general, las características clínicas no son evidentes al nacimiento. Durante la lactancia y la infancia, van apareciendo: la talla baja, la displasia ósea, engrosamiento de las válvulas cardiacas, síndrome del túnel del carpo, retraso mental, hepato y esplenomegalia, el hirsutismo, un desarrollo anormal y retraso psicomotor4.
Además, la característica facies tosca, con labios gruesos, boca abierta y puente nasal aplanado ayudan a hacer el diagnóstico.
Dependiendo de la forma, el retraso mental se manifiesta generalmente durante los primeros años de la vida. También puede ayudar la historia familiar.
Es posible hacer el diagnóstico prenatal mediante la determinación de la actividad enzimática en el cultivo de células del líquido amniótico o en las muestras de biopsias de vellosidades coriales. Como los resultados falsos, tanto positivos como negativos, son frecuentes, es necesario tener cuidado al interpretar las pruebas de detección sistemática postnatales efectuadas en la orina. Incluso en los pacientes gravemente afectados, las pruebas pueden ser negativas durante las primeras etapas de la lactancia. El diagnóstico se confirma analizando la actividad enzimática específica en los linfocitos, en los cultivos de fibroblastos o, en algunos tipos, en el suero4.
Las alteraciones radiológicas del esqueleto son típicas de una disostosis múltiple y, aunque su gravedad varía con los tipos, suelen ser lo bastante específicas como para permitir el diagnóstico preciso. No obstante, estas manifestaciones radiológicas cambian considerablemente durante la infancia y su interpretación ha de hacerse con gran cuidado4.
PRESENTACIÓN DEL CASO
Paciente de sexo masculino de 7 años de edad, procedente de Cochabamba, ingresa al Hospital Obrero N°2, Servicio de Pediatría, presenta un cuadro clínico de tres días de evolución caracterizado por alzas térmicas de 39 °C y tos que aumentaba en frecuencia, con diagnóstico de base de síndrome de Hunter, que fue diagnosticado al año y seis meses de vida.
Antecedentes de importancia: dos episodios de crisis convulsiva e internaciones reiteradas por bronco neumonía, faringitis aguda y retardo en el desarrollo psicomotor.
Al examen físico el paciente se encuentra en regular estado, afebril, normo hidratado, con signos vitales de FC 126/min, FR 22/ min, PA 110/90 mmHg, T 37.5°. Cabeza: macrocefalia, asimétrica, a la palpación presencia de masas en sentido horizontal de consistencia dura en los parietales, no dolorosas (ver figura 1), Cuello: corto. Fascie: mongólica, característica de su enfermedad. Nariz: en silla de montar. Oídos: hipoacusia bilateral. Boca: afasia, macroglosia, amígdalas hipertróficas. Abdomen: globoso, hepato y esplenomegalia. Tono y trofismo: piel gruesa y disminuida. Pulmones: estertores, crepitos y sibilancias en ambos campos pulmonares. Corazón: ruidos sobre agregados por patología pulmonar.
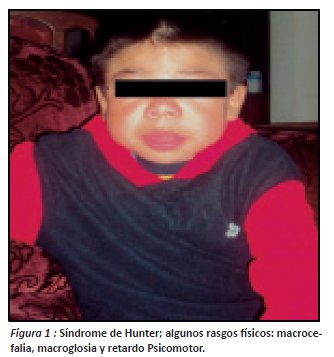
Extremidades superiores: mano en garra (fig. 2).

Examen neurológico: retardo psicomotor y retardo mental progresivo.
A la impresión diagnóstica: bronconeumonía, síndrome bronquial obstructivo, retardo del desarrollo psico motor y síndrome de Hunter.
La madre refiere que realizó un viaje al exterior del pais, donde llegaron al diagnóstico de Síndrome de Hunter.
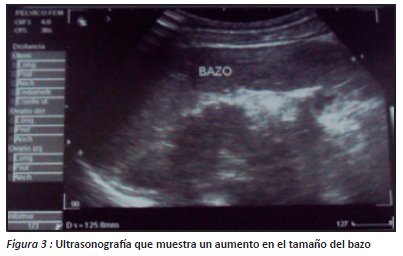
En el servicio de pediatría del hospital obrero N° 2 se solicitaron los siguientes exámenes complementarios: Placa radiográfica de tórax: donde se presencia aumento del trama bronco vascular e infiltrados parahiliares bilaterales y Ecografía abdominal: que mostró al Hígado: de forma y contorno normal, aumento de tamaño cuyo borde anterior llega hasta la línea umbilical, eco estructura y ecogenecidad del parénquima normal y el Bazo: con ecogenecidad, eco estructura normal y aumento de tamaño, cuyo borde inferior cubre el polo inferior del riñón, longitud 126 mm (figura 3).
El diagnóstico diferencial es realizado con los otros tipos de mucopolisacaridosis, y eso se obtiene determinando la enzima y el glucosaminoglucano afectado. En el presente caso se determinó la ausencia de iduronato -2- sulfatasa y los (GAG) afectados son Dermatan Sulfato y Heparan sulfato.
Dada la circunstancia de la enfermedad y la ausencia de un tratamiento realmente eficaz, es importante recalcar la necesidad de un "tratamiento paliativo", contra los diversos síntomas. Su objetivo es reducir los efectos del deterioro de las funciones corporales.
En este caso resolver el problema pulmonar es crucial, ya que la principal causa de muerte de estos pacientes es por "problemas pulmonares y cardiacos".
Por la recurrencia de problemas pulmonares, se usaron antibióticos, el tratamiento médico fue cloranfenicol 100 mg/kg/día cada 6 horas, cloxacilina 100 mg/kg /día cada 6 horas por dos días, Dipirona 10 mg endovenosa cada 12 horas, carabamazepina 12,5 mg /kg /día vía oral cada 8 horas, disperidona 0,25 mg vía oral cada noche.
DISCUSIÓN
Si bien es poco frecuente esta patología, es muy importante conocerla, para poder hacer un buen diagnóstico temprano y saber clasificarla de las otras mucopolisacaridosis, además no hay que obviar que una sola patología puede afectar a diferentes órganos y lo más importante la repercusión que tiene en el sistema nervioso llevando a un deterioro progresivo de las funciones motoras y de la capacidad mental. No hay que olvidar que muchos de estos pacientes no llegan a vivir hasta los 20 años.
Es importante ver el contorno familiar, ya que tener un niño en este estado, produce un gran estrés para los padres y familiares, porque estos niños son de sumo cuidado y tienen que estar bajo supervisión las 24 hrs., sería muy importante el soporte psicológico en esta área.
La ausencia de tratamientos para la mayor parte de las MPS y el alto costo de la terapia de reemplazo enzimático para las formas menos severas de la MPS I han motivado a los investigadores a seguir desarrollando nuevas alternativas terapéuticas, entre ellas las terapias celulares, génicas y de inhibición de la síntesis de los sustratos depositados5.
Actualmente, se encuentran en desarrollo los estudios clínicos para probar la terapia de reemplazo enzimático en las MPS II y VI. Los resultados hasta ahora favorables hacen prever que muy pronto se dispondrá de un tratamiento específico también para estas últimas afecciones6.
Mientras que el tratamiento puede detener muchos de los síntomas, es ineficaz con los neurológicos, de modo que aunque aumenta la expectativa y la calidad de vida, no resuelve las deficiencias mentales.
Además el papel de la Consejería Genética juega un papel muy importante, hoy en día debería ser imprescindible en parejas con antecedentes de familiares con enfermedades genéticas. Es posible diagnosticar las portadoras mediante medición de la actividad enzimática en MPS tipo II, cuya herencia es recesiva ligada con el cromosoma X, al igual que hacer el diagnóstico prenatal mediante la medición de la actividad enzimática en las vellosidades coriónicas y cultivo de células de líquido amniótico3.
AGRADECIMIENTOS
Quisiera agradecer a los familiares del paciente por haber permitido la presentación del caso clínico, desearles todo lo mejor en sus actividades que realizan.
REFERENCIAS
1. Shire. Mucopolisacaridosis tipo II.. Pharmaceutica .México . SA. De CV; 2008 : 4-31. Disponible en: http://www.shire.com.br/attachments/0S2_folheto%20pacíente%20hunter.pdf [ Links ]
2. Maceira R MC, Atienza M G. Detección precoz de mucopolisacaridosis y oligosacaridosis en el período neonatal mediante cribado poblacional. Revisión sistemática [ Links ]
3. correa LN. Mucopolisacaridosis. Disponible en: http://www.scp.com.co/precop/precop _fíles/modulo_4_vin_3/mucopolisacaridosis.pdf [ Links ]
4. Manual de Merck . cap.269 Trastornos Endocrinos y Metabolicos. 10 ED. Editorial Harcourt SA. 1999. 153.
5. Mabe P. las Mucopolisacaridosis . Revista Chilena De Nutrición Scielo .vol. 31 (1); 2003 : 8-5. [ Links ]
Chaves A, López M. Mucopolisacaridosis y Guía de Familias.Coleccion Manuales y guía .2002. Nº 7 : 7 - 51. Disponible en :http://sid.usal.es/idocs/F8/8.1-6346/MUCOPOLISACARIDOSIS.pdf [ Links ]
6. ErosteguiC , Zalles L, Davalos R, Gamarra ML. Hidratos de Carbono .En : Bioquímica. 3 ra. Edición .CBBA: Editorial Metropolitana ; 2006: 53-55 [ Links ]

