











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La poroqueratosis es un trastorno poco frecuente de la queritinización de etiopatogenia desconocida.(1) Se considera un trastorno hereditario o adquirido, caracterizado clínicamente por la presencia de placas anulares con borde hiperqueratósico.(2)
Esta entidad fue identificada por primera vez por Neumann en 1875. No obstante, el término “poroqueratosis” fue introducido por el dermatólogo italiano Mibelli, quien lo utilizó para describir la afección observada en su paciente, destacando la implicación de los ostium ecrinos. Posteriormente, en 1893 Respighi y en 1937 Andrews describieron una forma superficial y diseminada de la enfermedad.(1)
Existen diversas variantes clínicas de poroqueratosis, entre ellas la poroqueratosis de Mibelli, la poroqueratosis actínica superficial diseminada, la forma lineal, la poroqueratosis palmaris et plantaris diseminada y la poroqueratosis punctata. Estas formas pueden diferir en su patrón de distribución, edad de inicio y asociación con factores desencadenantes como la exposición solar, inmunosupresión o traumatismos cutáneos.(3)
Muchos autores la consideran una entidad premaligna debido a su potencial degeneración hacia carcinoma epidermoide o basocelular, especialmente en lesiones crónicas o fotoexpuestas.(3) Por ello, el diagnóstico temprano y el seguimiento clínico son fundamentales.
En el presente artículo se expone un caso de poroqueratosis actínica superficial diseminada, destacando sus características clínicas, dermatoscópicas, hallazgos histopatológicos y enfoque terapéutico, con el objetivo de contribuir al conocimiento y reconocimiento de esta entidad infrecuente.
PRESENTACIÓN DEL CASO
Se presenta el caso de una paciente femenina de 79 años de edad, procedente de la ciudad de La Paz, Bolivia, de ocupación vendedora, con antecedente de exposición solar prolongada por más de 20 años. No refiere factores desencadenantes específicos. Como antecedentes patológicos, presenta diabetes mellitus tipo 2 controlada y enfermedad renal crónica en estadio G2, en tratamiento médico regular.
Consulta por un cuadro clínico de dos años de evolución caracterizado por la aparición progresiva de lesiones cutaneas hiperpigmentadas en la región facial, con incremento gradual en número y extensión acompañado de escozor ocacional. Al examen físico dermatológico se observan en la región frontal, mejillas, eminencias malares y mentón múltiples máculas pigmentadas de bordes figurados bien definidos, de color marrón oscuro con centro marrón claro, de tamaño variable entre 0,5 y 1 cm de diámetro (Imagen N°1,2,3); de distribución simétrica en áreas fotoexpuestas, en menor número en la región mandibular; similares lesiones se localizan en el dorso de ambas manos. A la palpación, levemente rugosas en su periferia.
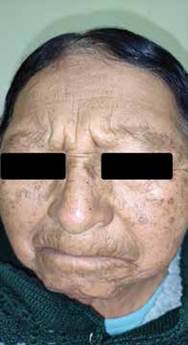
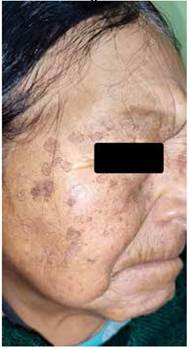
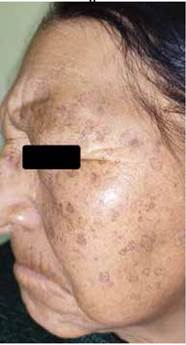
Al examen dermatoscópico: se evidencia máculas pigmentadas bien delimitadas de borde queratócico con puntos negros periféricos (trail tracks) y aclaramiento central (Figuras N°1 y 2).
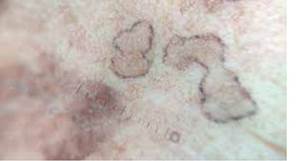

Se realizó biopsia cutánea incisional, cuyo estudio histopatológico fue concluyente con poroqueratosis actínica superficial diseminada, evidenciándose la presencia de una lámina cornoide característica(Figuras N° 3 y 4).
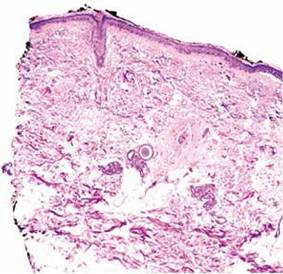
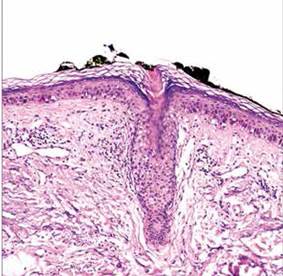
Figura N° 3. Tejido cutáneo con atrofia epidérmica, estrato basal con aumento de melanina y reacción de interfase vacuolar, dermis con elastosis solar difusa. Figura N°4. Se evidencia epidermis con paraqueratosis (lámina cornoide) dentro de una invaginación epidérmica, dermis con infiltrado infamatorio crónico.
DISCUSIÓN
Las poroqueratosis constituyen un grupo poco frecuente de dermatosis, ya sean de origen adquirido o hereditario, cuya causa exacta aún no se ha determinado y que se distinguen por presentar una alteración en el proceso de queratinización epidérmica.(3)
La poroqueratosis se clasifica principalmente en formas localizadas y generalizadas. Como se muestra en Tabla N°1(1):
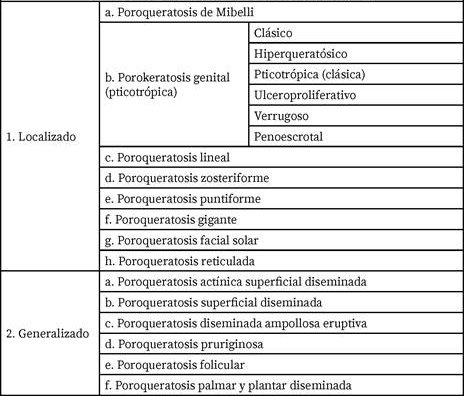
Las variantes localizadas más comunes son la poroqueratosis clásica de Mibelli, la poroqueratosis lineal, la poroqueratosis puntiforme, la poroqueratosis facial solar y la poroqueratosis genital. Y en relación a las variantes generalizadas están la poroqueratosis superficial diseminada, la poroqueratosis actínica superficial diseminada y la poroqueratosis palmoplantar diseminada.(1)
La poroqueratosis actínica superficial diseminada (PASD) es una genodermatosis con baja incidencia.(4) En relación a su epidemiología usualmente ha sido reportado en países con alta exposición solar, generalmente se presenta en adultos jóvenes o de mediana edad, con un pico de incidencia entre la tercera y cuarta década de la vida; sin embargo, también se han documentado casos de aparición tardía,(5) como el de nuestra paciente, quien desarrolló las lesiones en la séptima década.
Desde el punto de vista clínico se caracteriza por la aparición de múltiples máculas, pápulas y/o placas anulares distribuidas de forma bilateral y simétrica en áreas fotoexpuestas, especialmente en las extremidades distales. Estas lesiones suelen presentar una tonalidad eritematosa o parduzca, con un borde hiperqueratósico más prominente que el centro atrófico, lo que les confiere un aspecto anular o de contorno irregular debido a su crecimiento centrífugo progresivo(4),(6).
En la actualidad, se ha reconocido una variante denominada poroqueratosis actínica superficial diseminada pigmentada (PASDP), considerada una forma pigmentada de la PASD clásica. Esta se caracteriza por la presencia de un borde hiperpigmentado bien definido y por la incontinencia de pigmento en la dermis superficial, hallazgo que explica su tonalidad más oscura.(7) La PASDP se observa con mayor frecuencia en individuos de piel más oscura, como en el caso de nuestra paciente.
La afectación facial es menos habitual, reportándose solo en un pequeño porcentaje de casos (aproximadamente el 15%), generalmente limitada a las mejillas.(5) En el caso descrito, las lesiones se localizaron predominantemente en la región facial, una presentación infrecuente que resalta la variabilidad clínica de esta entidad.
La patogenia de la poroqueratosis no se comprende por completo, pero se considera el resultado de una clonación anómala de queratinocitos que conduce a una alteración focal de la queratinización epidérmica, estas células mutadas originan la lamela cornoide característica(2). Entre los factores implicados se ha descrito un patrón de herencia autosómico dominante con penetrancia variable, aunque en algunos casos se han identificado mutaciones somáticas como causa esporádica. Se han localizado seis loci genéticos asociados en los cromosomas 1, 12, 15, 16 y 18;(3),(8). Se asocia con mutaciones en el gen mevalonato quinasa ubicadas en los cromosomas 12q24 y 15q24.(18)
Además, estudios recientes han identificado mutaciones en genes de la vía del mevalonato, implicados en la síntesis de colesterol y la diferenciación epidérmica, lo que sugiere un papel importante en la alteración de la queratinización(9). Otros genes potencialmente relacionados con la diferenciación epidérmica anómala incluyen SSH1, SART3 y SLC17A9(8).
Entre los factores ambientales, la radiación ultravioleta (UV) desempeña un papel determinante, dado que múltiples estudios han demostrado la predilección de las lesiones en áreas fotoexpuestas y su exacerbación tras la exposición solar, lo que refuerza la influencia del daño actínico acumulativo en la expresión clínica de la enfermedad.(3)
Asimismo, la enfermedad se ha vinculado con estados de inmunosupresión, aunque el mecanismo exacto aún no ha sido aclarado.(3)) Algunos estudios sugieren una alteración en la respuesta inmunitaria cutánea, evidenciada por un aumento de la expresión de antígenos HLA- DR en las células de Langerhans epidérmicas.(10)) También, se han descrito casos relacionados con el uso de diversos fármacos (como suramina, hidroclorotiazida, furosemida, hidroxiurea, gentamicina, exemestano, flucloxacilina y agentes biológicos como etanercept, certolizumab y trastuzumab),(3) así como con enfermedades crónicas sistémicas, entre ellas enfermedad de Crohn, hepatopatía crónica, insuficiencia renal, amiloidosis, pancreatitis, síndrome de Sjögren, artritis reumatoide, espondilitis anquilosante, miastenia gravis y diabetes mellitus.
El diagnóstico de la poroqueratosis actínica superficial diseminada es fundamentalmente clínico, basado en la morfología típica de las lesiones anulares con borde hiperqueratósico. A la dermatoscopia destaca la presencia de borde periférico blanco (líneas del cráter volcánico), un área central blanca homogénea similar a una cicatriz, glóbulos o puntos marrones, estructuras vasculares (vasos sanguíneos puntuales, vasos lineales irregulares que cruzan la lesión), además la dermatoscopia UV resalta el borde hiperqueratósico, que brilla como un collar de diamantes.(11)
En la poroqueratosis actínica superficial diseminada (DSAP) pigmentada se observa la presencia de máculas hiperpigmentadas circulares bien delimitadas, que pueden mostrar puntos o líneas negras periféricas, conocidas como “trail tracks”. Estas corresponden al borde hiperqueratósico característico de la lesión. El centro suele presentar un aclaramiento progresivo, sin evidenciar características melanocíticas.(7) Este patrón clínico y dermatoscópico coincide con el observado en nuestra paciente.
El estudio histopatológico confirma el diagnostico, caracterizándose por la presencia de la lámina cornoide, una delgada columna de células paraqueratósicas dispuestas verticalmente sobre una depresión epidérmica, con ausencia o disminución de la capa granulosa y queratinocitos disqueratósicos o vacuolizados en la capa espinosa. En la dermis superior se observa un infiltrado inflamatorio linfocítico CD4+ de intensidad variable, a veces con características liquenoides, junto a vasos dilatados y ocasionales depósitos de amiloide.(5)) Estos hallazgos concuerdan con los observados en la biopsia de nuestra paciente, confirmando el diagnóstico de PASD.
El tratamiento de la poroqueratosis incluye diversas modalidades tópicas, sistémicas y físicas. Sin embargo, existe pocos datos de ensayos clínicos que orienten de manera efectiva el tratamiento.(12)
Entre las opciones tópicas se incluyen fluorouracilo 5%, imiquimod, tretinoína (0,05-1%), calcipotriol, diclofenaco 3%, y combinaciones de simvastatina o lovastatina 2% con crema de colesterol 2%, así como crisaborol 2%. Las terapias quirúrgicas y destructivas comprenden crioterapia, curetaje, electrodesecación, escisión quirúrgica y dermoabrasión. El empleo de láseres fraccionados, Q-switched, Nd:YAG y ER:YAG ha mostrado mejoría en las lesiones, aunque generalmente sin alcanzar resolución completa, similar a lo observado con la terapia fotodinámica.(13) Las terapias sistémicas se utilizan con menor frecuencia e incluyen retinoides como acitretina o isotretinoína, con respuestas variables.(14),(15) Recientemente, se ha reportado que la palimerfina logró la eliminación sostenida de las lesiones a los 12 meses en un paciente, empleada como estrategia profiláctica frente a mucositis.(16)
Sin tratamiento, las lesiones suelen permanecer de forma indefinida.(13)
En el caso de la paciente reportada, se indicó el uso de corticoide tópico (Hidrocortisona al 1%) para el manejo del prurito, medidas de prevención de fotoprotección y educación sobre signos de alerta para cáncer de piel.
Finalmente, la asociación entre la poroqueratosis y el desarrollo de neoplasias cutáneas como el carcinoma basocelular (CBC), el carcinoma epidermoide (CEC) o, con menor frecuencia, el melanoma maligno (MM) se estima en aproximadamente 3,4%. Este potencial de transformación maligna podría estar vinculado a alteraciones en las vías de señalización MAPK y fosfatidilinositol-3-quinasa/AKT, así como a mutaciones en el gen supresor tumoral p53, implicadas en la proliferación y supervivencia celular anómala.(17)
CONCLUSIÓN
La poroqueratosis actínica superficial diseminada (PASD) pigmentada es una entidad poco frecuente dentro de las poroqueratosis, se caracteriza por una alteración focal de la queratinización epidérmica asociada a factores genéticos, ambientales e inmunológicos. Su reconocimiento clínico y dermatoscópico resulta esencial para el diagnóstico oportuno y la diferenciación de otras dermatosis pigmentadas. Si bien su evolución es crónica y benigna, existe un riesgo de transformación maligna, por lo que la vigilancia dermatológica periódica y la educación del paciente en fotoprotección son fundamentales. El caso presentado resalta la importancia de considerar esta entidad en pacientes de edad avanzada y fototipos oscuros con lesiones faciales pigmentadas atípicas.
En el caso presentado, es importante considerar que la exposición solar crónica sin protección adecuada ligada a una actividad laboral (vendedora en la calle) durante mas de treinta años y la asociación a predisposición genética son factores de riesgo para la aparición de esta enfermedad poco frecuente.

