











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCION
Las vasculitis asociadas a anticuerpos anti citoplasma de neutrófilos (ANCA) son un grupo de trastornos que involucran vasculitis sistémica grave de vasos pequeños y se caracterizan por el desarrollo de auto anticuerpos contra las proteínas de los neutrófilos proteinasa 3 (PR3- ANCA) o mieloperoxidasa (MPO-ANCA). Los tres subgrupos de vasculitis a saber, granulomatosis con poliangeítis (GPA), poliangeítis microscópica y eosinofílica (EGPA), se definen según las características clínicas. Sin embargo, los hallazgos clínicos y genéticos sugieren que estos síndromes clínicos pueden clasificarse mejor como vasculitis positiva para PR3 (PR3-AAV), vasculitis positiva para MPO (MPO-AAV)(1).
La granulomatosis con poliangeítis es un trastorno poco común, con una incidencia anual reportada de 8- 10 casos por un millón de personas, la enfermedad puede aparecer a cualquier edad, en promedio 15% de los pacientes tiene <19 años de vida y la proporción de sexo es de varones/mujeres 1:1. Su causa es desconocida y se encuentra asociada con anticuerpos anticitoplasma de neutrófilo (ANCA) (2). No contamos con un estudio de prevalencia en nuestro medio, sin embargo, en el ambiente hospitalario, es frecuente observar un predominio mayor en esta condición a diferencia de sus otras variantes como poliangeítis microscópica y eosinofílica.
Es común que se encuentren 1 o más enfermedades autoinmunes diagnosticadas en un mismo paciente, sin embargo, en este apartado observaremos una asociación poco frecuente, entre vasculitis e inmunodeficiencia primaria.
CASO CLINICO
Ingresa a nuestro nosocomio paciente masculino de 50 años de edad, nativo de Bolivia - La Paz, fumador, con Índice tabáquico: 20. Tiene antecedentes de Granulomatosis con poliangeitis desde el 2021, tratada inicialmente con inmunomoduladores, (cilofosfamida), y anticuerpos monoclonales (rituximab). Presento episodios de neumonías en los últimos dos años. No se realizó cirugías y niega alergias. Este refiere disfonía, dificultad respiratoria moderada, tos productiva purulenta con episodios hemoptoicos y alzas térmicas no cuantificadas desde hace 6 días.
Se verificaron los siguientes signos vitales a su ingreso; presión arterial: 100/60 mmHg, frecuencia cardiaca 98 lpm, frecuencia respiratoria: 21cpm, saturación arterial de oxigeno: 82% con una fracción de inspiración de oxigeno: 0.21. Al examen físico cuello corto con acrocordones en región cervical, a nivel pulmonar con estertores crepitantes difusos en ambos hemitórax y soplo tubarico en región interescapular derecha. En la biometría hemática destaca hemoglobina de 10.6g/dl, leucocitos de 15400 uL, segmentados de 92%, plaquetas 270000 UL, Cuenta con química sanguínea con creatinina 2.1 mg/dl y perfil inmunológico ANCA P y ANCA C son positivos, destaca IgG encontrándose en niveles superiores en dos muestras repetidas. Además, cuenta con tomografía simple de tórax donde se observa en el corte axial en ápices pulmonares múltiples cavernas de bordes irregulares de hasta 9 cm, con niveles hidroaereos, rodeadas de focos de consolidación. (FIGURA 1)
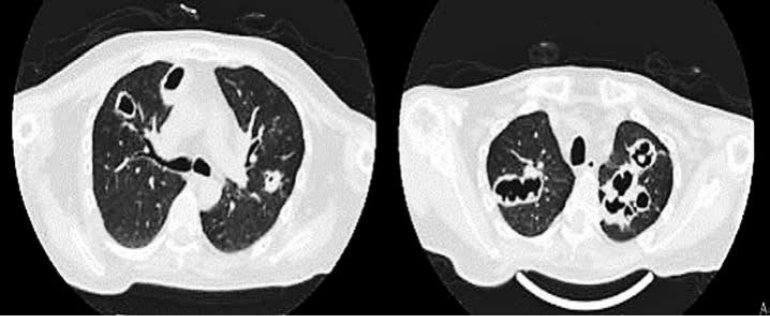
Inicialmente se inició tratamiento antibiótico empírico para neumonía atípica. Al obtener un rescate de Klebsiella oxitoca, sensible a imipenem por medio de lavado broncoalveolar, se instauro dicho régimen de tratamiento sin mejoría clínica significativa, se consideró descartar otras infecciones, en este caso, oportunistas por antecedente de tratamiento inmunosupresor, sin embargo, las pruebas fueron negativas incluyendo cultivo BK y Gen-Xpert para Micobacterium tuberculosis. Por mala evolución clínica, se planteó la posibilidad de un síndrome de inmunodeficiencia asociada al cuadro clínico actual. Por lo que se solicitó nuevamente un perfil inmunológico con cuantificación de inmunoglobulinas con la determinación de IgG especificas (IgG 1, IgG2, IgG 3 e IgG4) donde se observa deficiencia de IgG subclase-3 y serología para VIH negativa.
Se prescribe tratamiento inmunomodulador (gammaglobulinas) a dosis de 0,4 g/kg/dia por 5 días consecutivos. Paciente a la semana de inicio de gammaglobulinas, presento mejoría clínica significativa, con remisión progresiva de dificultad respiratoria por lo que se dio de alta para continuar tratamiento ambulatorio. Acudió al centro hospitalario después de un mes de tratamiento donde obtenemos un control tomografíco simple de tórax, en la que se observa campos pulmonares sin lesiones consolidativas en comparación con el estudio de ingreso. (FIGURA 2)
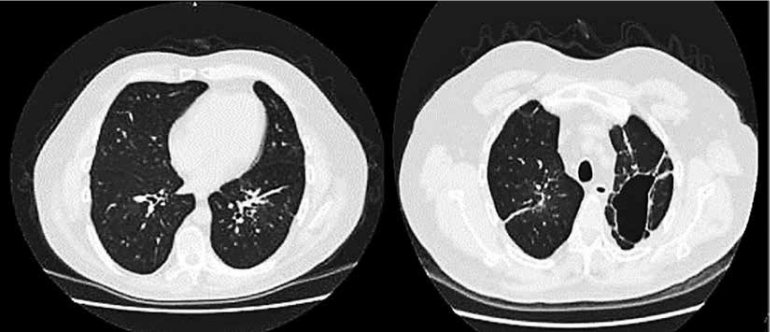
DISCUSION
Cada una de las cuatro subclases de IgG humanas exhibe un perfil único de funciones efectoras relevantes para la eliminación de microorganismos infecciosos. La cual varía con la naturaleza del antígeno, esto da como resultado que las respuestas de anticuerpos a ciertos antígenos sean predominantemente o exclusivamente de una sola subclase de IgG, por lo tanto, la incapacidad de producir anticuerpos del isotipo óptimamente protector puede dar como resultado un estado de inmunodeficiencia selectiva. Esto es, lo que particularmente sucedió en el presente caso clínico, donde a pesar de un tratamiento de amplio espectro, teniendo un rescate microbiológico especifico con descripción de sensibilidad al antibiótico administrado, no se observó mejoría clinica(3).
No se describen casos de inmunodeficiencia selectiva de IgG asociada a síndromes vasculiticos en la literatura en estos últimos años, sin embargo, se observaron un impacto en la mortalidad en pacientes hospitalizados con enfermedad pulmonar obstructiva crónica(4). En esta revisión realizada en el Hospital St. Paul, Vancouver, BC, Canadá, se observó que la mortalidad a un año fue del 56% en pacientes con deficiencia de IgG1, del 27% en deficiencia de IgG2, del 24% en deficiencia de IgG3 y del 31% en deficiencia de IgG4. Estos resultados concuerdan con la falta de respuesta inmunitaria humoral frente a infecciones bacterianas o micoticas, que están presentes también en pacientes con enfermedad pulmonar obstructiva y por la cual nos atreveríamos a comparar con las infecciones respiratorias en pacientes con granulomatosis con poliangeitis.
Los factores asociados con los niveles de IgG en adultos con deficiencia de subclase de IgG-3 no se comprenden completamente y en los adultos, la deficiencia de IgG3 puede ser la más común(5).
Las infecciones bacterianas del tracto respiratorio son el sello distintivo de las deficiencias primarias de anticuerpos(6) así como también de la granulomatosis con poliangeitis(2), de allí, que inicialmente no se halla sospechado de otra enfermedad concomitante en ese paciente, debido a que también se encuentran entre las infecciones más comunes en individuos sanos, las deficiencias primarias de anticuerpos suelen pasarse por alto en estos pacientes. Una evaluación cuidadosa de la historia, incluida la frecuencia, cronicidad y presencia de otras infecciones, ayudaría a sospechar deficiencia primaria de anticuerpos.
El tratamiento estándar para esta deficiencia de IgG es la terapia de reemplazo de inmunoglobulina(7,8), pueden prevenir infecciones y, por lo tanto, alterar el curso clínico de las deficiencias primarias de anticuerpos.

