











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO  uBio
uBio 
 Permalink
PermalinkINTRODUCCIÓN
El síndrome de Noonan (SN, OMIM 163950) es una de las enfermedades genéticas más comunes con una incidencia de 1/1000 a 1/2500 nacidos vivos1),(7. Tiene penetrancia completa y expresividad variable. Se engloba dentro un grupo de enfermedades conocidas como rasopatías, las cuales están causadas por mutaciones con ganancia de función en genes de la vía de señalización RAS/MAPK. Esta vía que está involucrada en importantes funciones biológicas como proliferación, supervivencia y diferenciación celular4),(5.
El SN clínicamente se caracteriza por presentar alteraciones craneofaciales heterogéneas; malformaciones cardiacas congénitas; talla baja y malformaciones torácicas (pectus carinatum superior, pectus excavatum inferior). Entre los signos clínicos menos frecuentes se encuentran: criptorquidia, escoliosis, pterigiium colli, defectos de la coagulación y defectos linfáticos en los pulmones, intestinos y/o extremidades inferiores; pérdida de la audición, retraso psicomotor, discapacidad intelectual, predisposición al cáncer1),(2),(4),(6),(8),(11).
Alrededor del 71% de los pacientes con síndrome de Noonan tienen mutaciones en alguno de los siguientes genes: PTPN11 (50 %), SOS1 (10- 15 %), RAF1 (5-15 %), RIT1 (4-9 %), KRAS (<5%), NRAS (<1 %), BRAF (<2 %), SHOC2 (<1 %),MEK1 (<1 %), CBL (<1 %), LZTR1(<1%) (4,12). En el 20-30% de los casos no se han identificado mutaciones causales4.
El diagnóstico molecular de síndrome de Noonan se establece mediante la identificación de una variante patogénica heterocigota en uno de los genes descritos o de una variante en homocigosis en el gen LZTR113.
El diagnóstico clínico se basa en los criterios de Van der Burgt, de acuerdo a estos criterios el diagnóstico se establece cuando se cumplen dos criterios mayores o un criterio mayor mas dos criterios menores o simplemente tres criterios menores6),(7),(14.
A continuación, se describe una serie de casos de cuatro pacientes no emparentados con diagnóstico clínico de síndrome de Noonan del Instituto de Genética de la Universidad Mayor de San Andrés durante la gestión 2017.
CASO 1
Paciente de sexo femenino de 1 año y 11 meses de edad, producto de primera gestación de padres no consanguíneos, nacida con peso 2,5 kg y talla 52cm (debajo del percentil 75th). Antecedentes patológicos: estenosis pulmonar. Al examen dismorfológico: Talla normal: 85cm (en percentil 50th). Ptosis palpebral derecha, frente prominente, micrognatia, pabellón auricular izquierdo con baja implantación y la derecha al límite (figuras 1 y 2). Pectum excavatum, cuello corto, pterigium colli, escoliosis dorsal izquierda, miembros inferiores con asimetría (derecho más largo que el izquierdo, 2 cm); hiperpigmentación: mancha oscura en brazo izquierdo y espalda, hipopigmentación en cara antero lateral del muslo derecho de 8cm aproximadamente. Prueba complementaria: Cariotipo 46, XX.
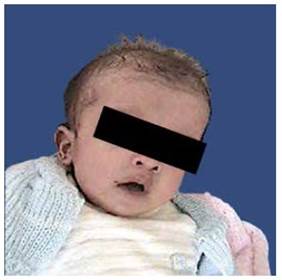
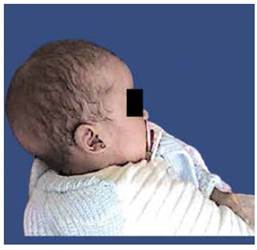
CASO 2
Paciente de sexo femenino de 3 años de edad, producto de tercera gestación de padres no consanguíneos, nacida con peso 3.1 Kg y talla 41 cm (debajo del percentil 5th). Antecedentes patológicos sin relevancia. Al examen dismorfológico: talla baja 85 cm (debajo del percentil 2th), Implantación capilar baja, ptosis palpebral derecha, paladar alto y ojival, cuello corto, tórax ancho con teletelia, soplo sistólico 4/6, abdomen globoso, cifoscoliosis con anomalía Sprengel en escapula derecha, braquidactilia, hiperlaxitud, genu valgo. Estudios complementarios: Cariotipo 46, XX; Ecocardiografía reporta estenosis valvular pulmonar e insuficiencia valvular pulmonar leve.
CASO 3
Paciente de sexo femenino de 15 años de edad, producto de la primera gestación de padres no consanguíneos (cariotipo de transmisión familiar: 45, XX,-13,-14,+t (13;14), sin relación con el síndrome de Noonan), nacida con peso: 3.6 Kg y talla: 49cm (en percentil 50th). Antecedentes patológicos: desnutrición crónica a los 20 días de nacida, dermatitis atópica a los 12 meses de edad, gastroenteritis a los 4 años, cirugía de ptosis palpebral izquierda a los 4 años y colitis a los 10 años. Antecedentes familiares: padre portador de translocación balanceada (13;14) y madre hipotiroidea. Examen dismorfológico: talla baja 116,5 cm (debajo del percentil 3th), facies asimétrica, pabellón auricular derecho con implantación al límite, pabellón auricular izquierdo con implantación normal, hélix superenrollado, fisuras palpebrales anti mongólicas, ptosis palpebral bilateral, nariz ancha, puente nasal bajo, narinas anchas, paladar alto, labios anchos, cuello corto, lesión cicatricial por hemangioma en labio mayor izquierdo, uñas de los dedos de la mano y los pies en vidrio de reloj, fuerza muscular 4/5 según la escala de Daniels y piel descamativa por dermatitis. Ecocardiografía: Estenosis pulmonar supra valvular.
CASO 4
Paciente de sexo femenino de 7 años de edad, producto de primera gestación de padres no consanguíneos, nacida con peso 2,6 Kg y talla 51 cm (en percentil 75th). Antecedentes patológicos: cirugía por comunicación interauricular acianógena y estenosis pulmonar valvular a los 4 años de edad. Antecedentes familiares, sin relevancia. Examen dismorfológico: talla baja 103cm (debajo del percentil 3th), braquicefalia, implantación baja de los dos pabellones auriculares con rotación posterior, lóbulos de las orejas pegados, anti hélix protruidos, fisuras palpebrales hacia abajo, ptosis palpebral izquierda, cejas con escasa cola, puente nasal bajo, narinas antevertidas, micrognatia, paladar ojival, hipertrofia de las amígdalas, tórax ancho, pectus carinatum, escoliosis dorsal, lordosis lumbar, anomalía de Sprengel, abdomen globoso, quinto dedo con braquidactilia, y pies equino varo. Se realiza cariotipo que reporta 46, XX.
DISCUSIÓN
El Síndrome de Noonan tiene un fenotipo variable, en parte debida a la heterogeneidad genética que presenta el síndrome, lo que dificulta el diagnóstico, sobre todo en edad adulta. Entre las características clínicas frecuentemente reportadas en la literatura se tienen: Facies dismórfica, talla baja, malformaciones torácicas, cardíacas y antecedentes de familiares diagnosticados4),(7),(11),(15),(16.
La facies se caracteriza por ptosis palpebral, hipertelorismo, orejas con rotación posterior e implantación baja, fisuras palpebrales oblicuas hacia abajo, frente prominente, cuello corto e implantación capilar posterior baja; este fenotipo varía con la edad, siendo más frecuente en edad temprana y no así en el adulto7),(11),(15),(16. En nuestra serie de casos además de las características faciales típicas descritas se observaron otras dismorfias faciales con diferente frecuencia de presentación: ptosis palpebral en el 100% de los casos; paladar ojival, cuello corto y rotación e implantación baja de orejas en el 75%; en el 50% de los casos se observó frente prominente, puente nasal bajo y ancho y micrognatia; en el 25% fisuras palpebrales oblicuas hacia abajo, implantación capilar baja y pterigium colli. Por otra parte el SN es considerado la segunda causa sindrómica de cardiopatía congénita, dentro de esta se describe la estenosis de válvula pulmonar con una frecuencia aproximada del 25-71%, seguida de defectos del septo atrial de 4-57% y cardiomiopatía hipertrófica del 10-29%5),(11),(13),(17. Los pacientes evaluados presentan estenosis de la válvula pulmonar en su totalidad, esto demuestra la importancia de descartar cardiopatía congénita en todo paciente con sospecha clínica de SN.
En cuanto a las otras manifestaciones descritas en literatura, la talla baja considerada como un criterio mayor, no se presenta en la totalidad de los casos, tres pacientes se encuentran por debajo del percentil 3, siendo lo esperable, sin embargo, una de las pacientes presenta talla normal. Esta situación coincide con reportes de la literatura donde se describe una frecuencia variable entre 50-100% de los casos de acuerdo al gen mutado13),(17.
En nuestra serie todos los pacientes representan casos de novo. La frecuencia de casos de novo en el SN varía entre el 30 al 75% según reportes de la literatura18.
Es interesante observar que existen otras características clínicas reportadas en la revisión de Kruszka et al., que no se encuentran en nuestra serie como hipotonía, dificultad de aprendizaje y/o discapacidad intelectual, malformaciones renales y hematomas11),(17.
Existen diversos síndromes con cierta superposición a las características clínicas del síndrome de Noonan, lo que hace esencial un diagnóstico diferencial meticuloso. Entre ellos, el síndrome de Turner, especialmente en individuos del sexo femenino, puede ser descartado mediante un cariotipo debido a su diferencia cromosómica, es decir, la presencia de un solo cromosoma X. En lo que respecta a las rasopatías, algunas se deben considerar en el diagnóstico diferencial. El síndrome de Noonan con múltiples léntigos (NSML) se destaca por el desarrollo de múltiples léntigos a lo largo de la vida del paciente, además de las características típicas del síndrome de Noonan. Por otro lado, el síndrome de Costello (CS) comparte varios rasgos con el síndrome de Noonan, tales como cardiopatías congénitas y baja estatura, pero a menudo presenta características cutáneas más marcadas, hipotonía y una mayor predisposición a ciertos tipos de cáncer. Además, los rasgos craneofaciales en el síndrome de Costello tienden a ser más toscos. El síndrome cardiofaciocutáneo (CFC) es una rasopatía rara que, si bien tiene similitudes con el síndrome de Noonan, como las anomalías faciales y cardíacas, también presenta una discapacidad intelectual de variada severidad y alteraciones cutáneas más frecuentes, incluyendo piel seca, escamosa o gruesa. El síndrome de Legius se caracteriza por macrocefalia y una capacidad neurocognitiva que varía desde normal hasta leve discapacidad intelectual. A diferencia de otros síndromes relacionados, no parece existir una predisposición aparente al desarrollo de cáncer en el síndrome de Legius. Finalmente, la neurofibromatosis-síndrome de Noonan (NFNS) presenta una combinación de características tanto de la neurofibromatosis tipo 1 como del síndrome de Noonan, incluyendo problemas de pigmentación en la piel y tumores benignos, así como los rasgos característicos de Noonan como la estatura baja y las anomalías cardíacas. Es esencial tener en cuenta todas estas condiciones al realizar un diagnóstico diferencial del síndrome de Noonan.7),(7),(14),(16),(19),(20.
En conclusión, el síndrome de Noonan, aunque genéticamente heterogéneo, presenta una constelación de manifestaciones clínicas consistentes que pueden guiar el diagnóstico y tratamiento del síndrome. Los individuos afectados pueden exhibir una serie de características distintivas, que incluyen baja estatura, cardiopatías, y manchas en la piel, además de otros posibles signos y síntomas. Es de vital importancia que los médicos y otros profesionales de la salud realicen una evaluación exhaustiva, a fin de no pasar por alto un diagnóstico potencial de síndrome de Noonan. Este artículo proporciona una visión global de las principales manifestaciones clínicas del síndrome de Noonan, y subraya la necesidad de considerar este diagnóstico en la evaluación de pacientes con las manifestaciones mencionadas.

