











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
El Virus Linfotrópico T Tipo 1 Humano (HTLV-1) es una infección desatendida y endémica en el mundo, causa mortalidad en el 5 a 10% de personas infectadas por no tener cura ni tampoco vacuna. Numerosos estudios han reportado focos de alta endemicidad en todos los continentes, destacando Australia, Japón, África Ecuatorial, América Latina 1. Las cifras mundiales son variables y oscilan desde 10 a 20 millones de infectados1,2,3.
La infección por HTLV-1 se asocia con una forma de cáncer de sangre (leucemia/linfoma de células T adultas), paraplejia de las piernas (paraparesia espástica tropical)4, artropatías, patologías como el lupus eritematoso sistémico y el síndrome de Sjögren5; estas enfermedades asociadas se generan solo en el 10% de infectados con el virus, mientras que el 90% permanecerán muchos años sin síntomas1,6, tal es así que, aún se desconoce los mecanismos virales y moleculares que favorecen al HTLV-1 a establecer una latencia libre de enfermedad durante años 7. El estudio de la interleucina-28B (IL28B) en HTLV-1 con variaciones genéticas en la respuesta inmune innata, podrían explicar el por qué muchos individuos infectados permanecen asintomáticos y otros desarrollan las enfermedades, mostrando entre ellos diferencias en la carga proviral, ya que se ha visualizado que algunos genotipos de IL28B, están asociados a una menor carga proviral y a una progresión clínica disminuida; esto podría sugerir que la progresión de la enfermedad puede estar favorecida por factores genéticos, de hecho se ha descrito que existe susceptibilidad genética para la infección por HTLV-1, pero esto necesita abordarse a profundidad8.
El gen IL28B ubicado en el cromosoma 19, codifica el interferón A3, una citocina que activa la vía JAK-STAT, induciendo así la transcripción de diversos genes estimulados por interferón (ISGs)3,9, que codifican proteínas con funciones antivirales 6, limitando así, la replicación viral a través del bloqueo de la traducción de proteínas virales e incremento de la degradación del ARN viral.
En este contexto, IL28B es una de las citocinas importantes la respuesta antiviral y se han caracterizado tres polimorfismos de un solo nucleótido (SNP) para este gen: rs12979860 (C > T); rs8099917 (T > G) y rs8103142 (T > C) 5. Los dos primeros SNPs han destacado debido a su impacto en la respuesta antiviral, asimismo, se ha demostrado que estos polimorfismos son fuertes predictores de respuesta viral sostenida o están asociados con la eliminación espontánea del virus de la hepatitis C (VHC)9,10, también se ha revelado que algunos alelos específicos se asocian con la falta de respuesta virológica en infectados con hepatitis B (VHB)11 y se ha investigado si estos mismos SNP tienen asociación con VIH/SIDA12.
Aun así, la información de la relación de los SNPs del gen IL28B (también IFNL3 y IFNL4) con el riesgo de infección por HTLV-1 es muy escasa y sigue sin ser aclarada. Los pocos estudios realizados de los SNPs rs12979860 y rs8099917 no son concluyentes o se centran en su asociación con manifestaciones clínicas asociadas a HTLV-15,13-15 llegando a tener incluso resultados controvertidos3.
Con la finalidad de buscar evidencia que muestre el rol de los polimorfismos en la susceptibilidad a la infección por HTLV- 1 y se contribuya al conocimiento de factores genéticos que puedan modular la respuesta antiviral, por lo tanto, el objetivo de este estudio fue realizar un metaanálisis para determinar la asociación entre los SNPs rs8099917 y rs12979860 del gen IL28B y el riesgo de infección por HTLV-1.
Material y métodos
Diseño del estudio y estrategia de identificación
Se realizó un metaanálisis de asociación genética siguiendo las directrices PRISMA, y su protocolo fue registrado en PROSPERO (código: CRD42025636396). Se efectuó una búsqueda minuciosa en tres bases de datos: Scopus, PubMed y Google Scholar. Las estrategias de búsqueda para cada base fueron las siguientes. Pubmed: ((“rs8099917” OR “rs12979860” OR “IL28B” OR “Interleukin 28B”) AND (“polymorphism” OR “SNP”)) AND (“HTLV” OR “Human T-cell lymphotropic virus”). Google Scholar: (“rs8099917” OR “rs12979860” OR “IL28B”) AND (“HTLV” OR “Human T-cell lymphotropic virus”) AND polymorphism. Scopus: TITLE-ABS-KEY ((“rs8099917” OR “rs12979860” OR “IL28B” OR “Interleukin 28B”) AND (“polymorphism” OR “SNP”) AND (“HTLV” OR “Human T-cell lymphotropic virus”)).
Únicamente se incluyeron estudios observacionales priorizando estudios de casos y controles y estudios transversales descriptivos que: (a) detallaron la frecuencia genotípica de los casos (infectados con HTLV-1) y los controles (sanos) para los SNPs rs8099917 y/o rs12979860; (b) permitan calcular OR (odds ratio) y (c) tener acceso al texto completo. Por lo escaso de la información no se consideraron criterios restrictivos relacionados con el año de publicación, ámbito geográfico o el idioma. Se excluyeron reportes experimentales en modelos animales, estudios in vitro sin datos poblacionales, asimismo para mantener la calidad y verificabilidad de los datos genotípicos reportados se excluyeron estudios provenientes de literatura gris, aquellos que no abordaron los SNPs de interés y aquellos que carecían de información sobre frecuencias genotípicas. La búsqueda se realizó entre diciembre de 2024 hasta enero de 2025.
Extracción de datos
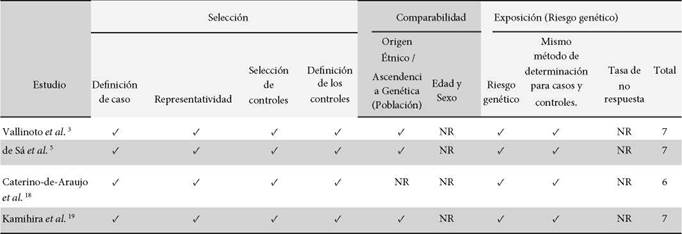
Una primera ronda de lectura de títulos y resúmenes se realizó para determinar si los estudios eran de interés para el metaanálisis; posteriormente los dos primeros autores realizaron una lectura íntegra de los estudios seleccionados. Todos los trabajos incluidos correspondieron a fuentes primarias, es decir artículos originales que reportaron datos experimentales o clínicos propios. De cada estudio se extrajo la información detallando: autores, año de publicación, SNP, tipo de infección (mono infección y/o coinfección), frecuencia genotípica del SNP en los casos (infectados) y controles (sanos). Las discrepancias de la inclusión se resolvieron por consenso. Se evaluó de manera independiente por uno de los autores, la calidad metodológica y riesgo de sesgo de los estudios incluidos usando la escala de Newcastle-Ottawa adaptada.
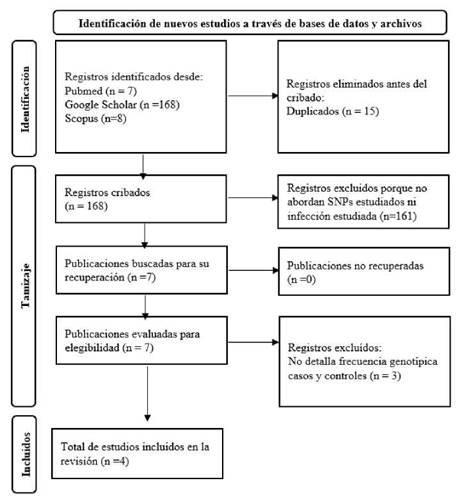
Metaanálisis
En una primera etapa se calculó el p-valor para el equilibrio de Hardy-Weinberg (HWE), se consideró que la población está en HWE cuando p >0,0516. Para establecer la asociación entre los SNPs rs8099917 y rs12979860 con la infección por HTLV-1 se calculó el OR y sus respectivos intervalos de confianza (IC al 95%). Cuatro modelos genéticos fueron analizados: alélico, recesivo, dominante y sobredominante. En todos los casos, se consideró como estadísticamente significativo un valor de p < 0,05. Para optar por un modelo de efectos fijos o aleatorios previamente se analizó la heterogeneidad (I2, valor Q y p- valor). Asimismo, se aplicó el test de Egger para evaluar el sesgo de publicación, considerándose p- valor de este test inferior a 0,05 como posible indicio de sesgo. La información recolectada fue seleccionada y analizada considerando los siguientes criterios: claridad en la descripción del diseño del estudio; definición explícita de los grupos de casos y controles; disponibilidad de datos completos sobre frecuencias genotípicas, cumplimiento del HWE y consistencia metodológica con los objetivos de este estudio. El análisis estadístico se realizó empleando el sistema MetaGenyo17.
Resultados
La Figura 1 presenta el flujograma para inclusión de estudios. Se identificaron 183 estudios producto de la estrategia de búsqueda; 15 artículos se eliminaron por ser duplicados y al realizar el cribado por títulos y resúmenes se excluyeron 161 publicaciones que no abordaban los SNPs de interés ni la infección por HTLV-1. En la fase de lectura de texto completo, se excluyeron 3 artículos por no detallar frecuencias genotípicas de los SNPs en la población estudiada. Solo 4 estudios cumplieron todos los criterios de inclusión 3,5,18,19. La muestra total analizada incluyó 204 casos y 671 controles para el SNP rs12979860 y 226 casos y 492 controles para el SNP rs8099917. Las frecuencias genotípicas se detallan en la Tabla 1. No se encontró desviación del HWE (p>0,05) para ninguno de los SNPs estudiados. La calidad metodológica y el riesgo de sesgo de los estudios incluidos se muestran en la Tabla suplementaria 1.
Tabla suplementaria 1. Calidad de los estudios incluidos según la escala de Newcastle-Ottawa modificada.
Respecto a las asociaciones genéticas, el metaanálisis no encontró una relación significativa entre el SNP rs12979860 y la infección por HTLV-1 en ninguno de los modelos genéticos evaluados (Figura 2): modelo alélico C vs T (OR = 0,98; p = 0,89); modelo recesivo CC vs CT+TT (OR = 1,03; p=0,85); modelo dominante CC+CT vs TT (OR = 0,91; p = 0,63); modelo sobre dominante CT vs CC+TT (OR = 0,92; p = 0,59).
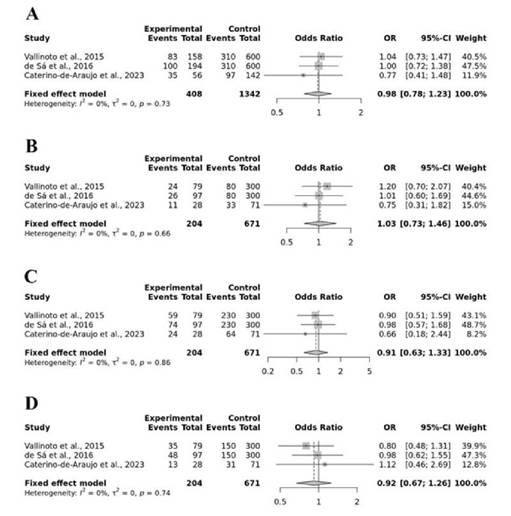
Fuente: Elaboración propia.
Nota: Los términos en inglés significan: “study” (estudio); “events” (eventos observados); “95%-CI” (intervalo de confianza al 95%); “weight” (peso).
Figura 2. Forest plot del metaanálisis para la asociación del SNP rs12979860 con el riesgo de infección por HTLV- 1. Se muestra el análisis para modelos (A) alélico C vs T, (B) recesivo CC vs CT+TT, (C) dominante CC+CT vs TT, (D) sobredominante CT vs CC+TT.
Tabla 1. Frecuencia genotípica de los SNPs rs12979860 y rs8099917 del gen IL28B en casos y controles de HTLV-1, se muestra el equilibrio de Hardy-Weinberg calculado
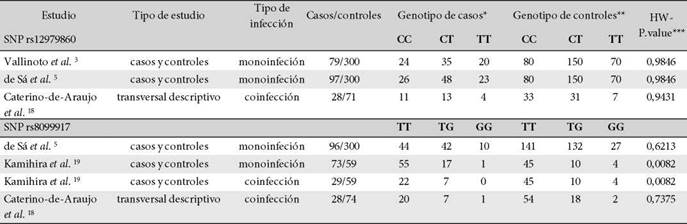
Fuente: Elaboración propia. Nota: * infectados con HTLV-1; ** sin infección; *** calculado por la prueba de chi-cuadrado.
De manera similar, el análisis del SNP rs8099917 tampoco mostró asociaciones significativas con la infección por HTLV-1 (Figura 3). Los valores de OR obtenidos no fueron significativos en todos los modelos analizados: modelo alélico T vs G (OR = 1,01; p=0,97); modelo recesivo TT vs TG+GG (OR = 0,95; p= 0,78); modelo dominante TT+TG vs GG (OR = 1,09; p=0,80); modelo sobre dominante TG vs TT+GG (OR = 1,12; p=0,53).
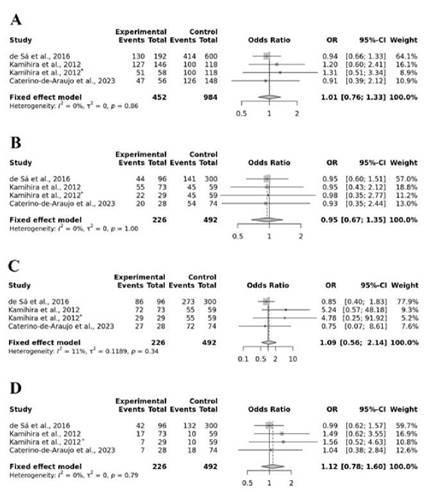
Fuente: Elaboración propia.
Nota: *Coinfección. Los términos en inglés significan: “study” (estudio); “events” (eventos observados); “95%-CI” (intervalo de confianza al 95%); “weight” (peso).
Figura 3. Forest plot para la asociación del SNP rs8099917 con el riesgo de infección por HTLV-1. Se muestra el análisis para modelos (A) alélico T vs G, (B) recesivo TT vs TG+GG, (C) dominante TT+TG vs GG, (D) sobredominante TG vs TT+GG.
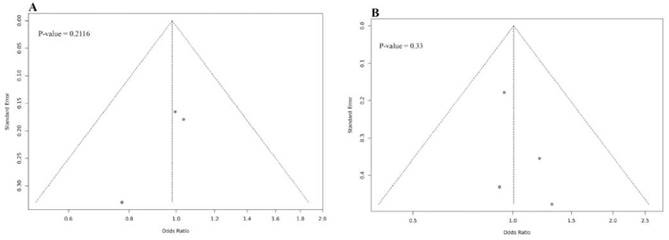
Fuente: Elaboración propia.
Nota: Los términos en inglés significan: “standard error” (error estándar); “p-value” (p valor).
Figura suplementaria 1. Gráficos de embudo para sesgo de publicación en estudios seleccionados. (A) rs12979860 y riesgo de infección con HTLV-1, modelo alélico C vs T. (B) rs8099917 y riesgo de infección con HTLV-1, modelo alélico T vs G.
En la Figura suplementaria 1 se presenta los resultados del análisis de sesgo de publicación utilizando gráficos de embudo y el test de Egger; en ambos SNPs no se observó asimetría en las gráficas. Las pruebas de Egger muestran que no existe sesgo de publicación para rs12979860 (modelo alélico C vs T, p= 0,2116) ni para rs8099917 (modelo alélico T vs G, p= 0,33). La prueba de Egger para los otros modelos genéticos indica también que no existe sesgo de publicación (p> 0,05).
Los valores numéricos de las Figuras 2, 3 y la Figura suplementaria 1 se presentan con notación decimal anglosajona (con punto) debido a la configuración del software empleado.
Discusión
Los SNPs del genIL28B, conocidos actualmente como IFNL3 e IFNL418,20,21, se han asociado con la susceptibilidad y progresión de distintas infecciones virales. Se ha descrito que en el caso de COVID-19, ciertos genotipos de rs8099917 y rs12979860 pueden influir en la resistencia a esta infección22; en el caso de VHB algunos se vinculan con el aclaramiento espontáneo y otros con la persistencia de la infección11, del mismo modo, estos SNPs actúan como factores de riesgo en infección por VHC23, y también se han estudiado en VIH 12 y citomegalovirus 24. Aunque Assone et al. (2 014) reporta que algunos SNPs de IL28B son más frecuentes en infectados con HTLV-1 13, su asociación con esta infección ha sido poco estudiada. De Sá et al. (2016), sugieren que rs12979860 y rs8099917 están relacionados con el desarrollo de artropatías, mielopatía y/o paraparesia espástica tropical en infectados con HTLV-15; asociándose incluso con mayores cargas provirales 13, particularmente en portadores con el genotipo CT/TT de rs1297986025, además, el genotipo CC de este mismo SNP podría actuar como posible factor agravante asociado a niveles elevados de TNF-β e IFN-γ5. Por otra parte, el genotipo GG de rs8099917 parece modular la respuesta inmune a HTLV-113 y el genotipo TT de rs8099917 podría ser un factor de riesgo para la elevación de la carga proviral5. Dado su potencial como biomarcador, algunos autores proponen investigar los polimorfismos deL gen IL28B para el seguimiento y monitoreo de personas infectadas con HTLV-13,13.
Los resultados del presente metaanálisis no respaldan que exista asociación de rs12979860, ni de rs8099917 con el riesgo de infección por HTLV-1 en ninguno de los modelos genéticos analizados (alélico, recesivo, dominante y sobredominante), aunque estos resultados parezcan inesperados hay evidencia que respaldan nuestros hallazgos; como lo encontrado por Kamihira et al. (2 012)19, quienes hallaron que las frecuencias de homocigotos TT para rs8099917 no están asociados a portadores de HTLV- 1 y por ende el SNP rs8099917 no está asociado con la susceptibilidad a la infección por HTLV-119; también Vallinoto et al. (2 015), reportaron que no existen diferencias de las frecuencias genotípicas de rs12979860 entre sintomáticos, asintomáticos y controles, y que rs12979860 no tiene influencia en la carga proviral de HTLV 3, además concordamos con Sanabani et al. (2 012)14, quienes tampoco encontraron evidencia que respalde que los SNPs del gen IL28 se comporten como marcadores de riesgo y/o susceptibilidad genética frente a la infección por HTLV-1 y plantean la posibilidad que los reportes previos de asociaciones de estos SNPs con HTLV-1 se deban al azar o las asociaciones se hayan exagerado debido tamaño reducido de la muestras en estos estudios previos.
Por otra parte, es pertinente indicar que, conociendo que el HWE es fundamental en el análisis de datos genéticos específicamente en estudios metaanalíticos 26, en nuestro estudio no observamos desviación del HWE (p>0,05) en los SNPs estudiados. Es decir, los genotipos de los controles cumplen con el HWE, lo cual se interpreta que los datos analizados no están afectados por errores de sesgo de selección, estratificación poblacional, ni por errores de genotipado; no obstante el cumplimiento del HWE por sí solo no descarta posibles errores sistemáticos en los estudios incluidos por eso solo debe interpretarse como indicador básico de consistencia genética, del mismo modo las pruebas de Egger muestran que no hubo sesgo de publicación en los artículos analizados.
Las principales limitaciones de este trabajo se relacionan con los pocos estudios incluidos en el metaanálisis, esto podría impactar en la producción de resultados sólidos. Existe la posibilidad que, debido al número reducido de estudios disponibles, el metaanálisis tenga bajo poder para detectar asociaciones genéticas débiles, asimismo respecto a las pruebas de Egger, dado que los estudios incluidos fueron menores a 10, estas pruebas tienen baja potencia en este contexto. Otra limitación es que los estudios incluidos en este metaanálisis solo corresponden a poblaciones de Brasil 4,5,18 y Japón 19, y se conoce que la frecuencia de SNPs puede diferir en distintas poblaciones y grupos étnicos. No fue posible evaluar interacciones gen-ambiente, tampoco efectos asociados a coinfecciones u otras variables clínicas, las cuales podrían modificar los resultados mostrados; debido a esto, no se puede descartar asociaciones en otros grupos étnicos o en distintos fenotipos clínicos.
Conclusión
A pesar de la importancia que tienen los perfiles genéticos en diversas infecciones virales, los resultados del presente metaanálisis nos permiten concluir que, los polimorfismos de un solo nucleótido rs8099917 y rs12979860 del gen IL28B, no están asociados al riesgo de infección por HTLV-1 en ninguno de los cuatro modelos genéticos que se han analizado. Debido a la escasez de estudios, la posible influencia de factores ambientales o variabilidad genética entre poblaciones, los hallazgos deben tomarse con precaución y consideramos que es necesario llevar a cabo estudios multicéntricos adicionales con muestras de gran tamaño, y diferentes grupos étnicos para poder contrastar y reforzar la evidencia actual.

