











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
La proteinosis alveolar pulmonar (PAP) es una enfermedad rara de tipo intersticial que afecta de manera amplia a los pulmones.
Se caracteriza por la acumulación de material lipídico-proteico derivado del surfactante pulmonar. Esta acumulación es el resultado de una disminución en la eficacia de los macrófagos alveolares para eliminar dicho material, provocando obstrucción en los alvéolos y alteraciones en la oxigenación2.
Se estima que cerca del 90% de los casos son de origen autoinmune, seguido por un 4% de PAP secundaria, un 1% de PAP congénita, y un 5% restante que se atribuye a un origen indeterminado3. A pesar de su baja incidencia, la PAP autoinmune se observa con mayor frecuencia en hombres (proporción 2 a 1), y en individuos de la tercera y quinta década de vida4.
La PAP puede aumentar la susceptibilidad a infecciones respiratorias debido a la acumulación de proteínas en los alvéolos, debilitando la respuesta inmunológica local para combatir eficazmente la invasión de patógenos5-7.
Diagnosticar la PAP sigue siendo un desafío, y generalmente de debe sospechar en aquellos pacientes con disnea crónica y progresiva. Los métodos diagnósticos incluyen tomografía computarizada de tórax en busca del patrón de “crazy paving”. La broncoscopía permite identificar una secreción lechosa-espumosa en el lavado broncoalveolar. Es esencial considerar otros diagnósticos diferenciales, como la exacerbación de la fibrosis pulmonar, las neumonías intersticiales, las neumonitis inducidas por fármacos y la neumonía por Pneumocystis jirovecii8.
Presentamos un caso de proteinosis alveolar pulmonar que plantea desafíos en su diagnóstico. Resaltamos la importancia de considerar la PAP como parte del diagnóstico diferencial en pacientes con disnea crónica, y destaca el papel crucial de la broncoscopía para un diagnóstico preciso.
Presentación del caso
Mujer de 41 años, agricultora y es originaria y residente de Ayacucho, Perú. No presenta antecedentes personales ni familiares de importancia. Refiere tos seca intermitente sin predominio horario durante los últimos 11 meses, tratada con sintomáticos y medicinas alternativas herbolaria locales. Cinco meses antes del ingreso, la tos se torna productiva, se agrega dolor torácico de leve intensidad y disnea de grandes a medianos esfuerzos. La tos y disnea comenzaron a intensificarse hace aproximadamente cuatro meses, lo que llevó a buscar atención médica en el primer nivel de atención en varias ocasiones. Durante estas consultas, recibió tratamientos empíricos, como antihistamínicos y antibioticoterapia, pero los síntomas respiratorios mejoraron solo parcialmente. Refiere una baja ponderal de 7 a 10 kg en el último año.
Hace aproximadamente un mes, es decir, un mes después de haber recibido su último tratamiento con antibióticos empíricos en el primer nivel de atención, la paciente presentó un empeoramiento de la disnea, incluso en reposo y con el habla entrecortada. Fue llevada al hospital de Ayacucho con insuficiencia respiratoria y sospecha de neumonía bacteriana. En este centro, recibió tratamiento con ceftriaxona 2 g endovenosa durante 7 días y oxigenoterapia con cánula binasal a 3-5 litros por minuto. El oxígeno fue retirado al completar los 7 días de tratamiento, tras lo cual fue dada de alta y derivada al consultorio de neumología para seguimiento.
En la evaluación física realizada en el consultorio de neumología, la paciente ingresó con una presión arterial de 100/60 mmHg, con una frecuencia cardiaca de 60 pulsaciones por minuto, una frecuencia respiratoria de 18 por minuto, una saturación de O2 91 % a FiO2 ambiental y una temperatura axilar 38,5 grados Celsius. En el examen respiratorio, se auscultó un frémito vocal reducido en ambos pulmones, así como una disminución del murmullo vesicular en las bases pulmonares y crepitaciones finas difusos en los campos mediales. El resto de la evaluación física no mostró anomalías. Tras esta evaluación, se decidió su hospitalización y se le realizaron los siguientes exámenes complementarios:
En las analíticas sanguínea de ingreso se observaron los siguientes valores: leucocitos 9,030 K/ul, segmentados 58 %, bandas 0 %, linfocitos 33 %, monocitos 6 %, eosinófilos 3 % y hemoglobina 15,5 gr/dl. El perfil bioquímico no mostró alteraciones. En los estudios inmunológicos, los niveles de C3 fueron de 102,18 mg/dl y los de C4 de 40,13 mg/dl, ambos dentro de rangos normales. Además, se obtuvieron resultados negativos para Anti CCP, ANCA-C (Anti PR3) y ANCA-P (Anti MPO), ANA, Factor reumatoide (FR) método látex, RPR y el test de ELISA para VIH.
Se realizó una gasometría arterial con un porcentaje fraccional de oxígeno inspirado de 21%, una pCO2 de 33 mmHg, una pO2 de 57 mmHg, una concentración de bicarbonato (HCO3) de 27,3 micromoles por litro y un A-aDO2 de 148,8.
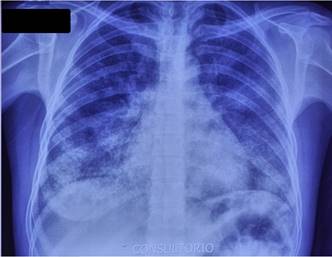
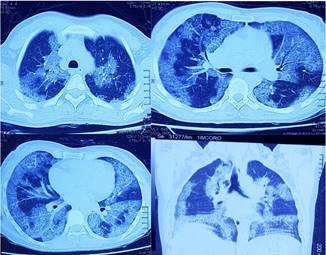
La radiografía torácica en proyección postero-anterior mostró infiltrados intersticio alveolar parahiliar y basal bilateral, con áreas de patrón alveolar bibasales en parches (Figura 1). Mientras que, la Tomografía de tórax mostró superposición de áreas con vidrio esmerilado y engrosamiento lineal de septos intra e interlobulillares con distribución parcheada y difusa, conocido como patrón “crazy-paving” (Figura 2).
Durante la broncoscopía, se observó una abundante secreción mucoide en todo el árbol bronquial, junto con un leve eritema en la mucosa. En el mismo procedimiento, se realizó un lavado broncoalveolar con suero fisiológico, obteniendo aproximadamente 70 ml de secreción lechosa, espumosa. No se identificaron anomalías endobronquiales. Además, se llevó a cabo una biopsia transbronquial de lóbulo medio e inferior derecho.
En el examen directo del esputo y del aspirado bronquial con la coloración de Ziehl-Neelsen, no se detectaron bacilos ácido-alcohol resistentes. La citología mostró la presencia de linfocitos y material eosinofílico proteico amorfo, junto con un componente inflamatorio.
El cultivo de gérmenes comunes en aspirado bronquial fue negativo. Además, el análisis anatomopatológico de la biopsia transbronquial reveló la presencia de numerosas células epiteliales del tracto respiratorio con macrófagos espumosos, que dieron positivo en la coloración ácido peryódico de Schiff (PAS), lo que indica cambios compatibles con proteinosis alveolar pulmonar (Figura 3).
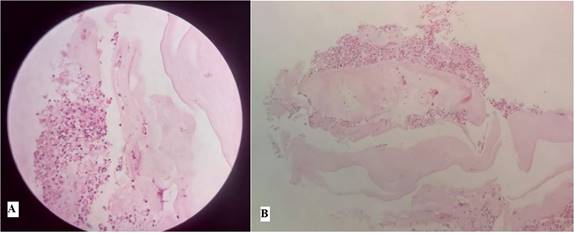
Figura 3. Estudio anatomopatológico de la biopsia transbronquial. Tinción hematoxilina eosina. A. Aumento 10X. B. Aumento 40X. Muestra constituida por material amorfo de eosinófilo con componente inflamatorio.
Los resultados de la espirometría, realizada al sexto día de hospitalización, revelaron una capacidad vital forzada (FVC) de 2,11 litros (69%), un volumen espiratorio forzado en el primer segundo (VEF1) de 1,94 litros (77%), y un índice FEV1/FVC de 91,9 %. Además, el flujo espiratorio máximo (PEF) fue de 3,72 litros (62%) y el flujo espiratorio forzado correspondiente al rango del 25% al 75% de la capacidad vital forzada (FEF 25-75) alcanzó 2,83 litros (97%). Estos hallazgos son compatibles con un sugerente de restricción leve, sin respuesta significativa al broncodilatador.
Durante su hospitalización, la paciente recibió oxigenoterapia con cánula binasal a un flujo de 2-3 litros por minuto, lo que ayudó a mantener una saturación de oxígeno adecuada. En cuanto al tratamiento sintomático, se le administró dextrometorfano como antitusígeno inicial, pero debido a la falta de respuesta, se optó por codeína, que mostró mayor eficacia en el control de la tos. Este manejo se implementó para aliviar los síntomas respiratorios. La paciente fue dada de alta tras 7 días de hospitalización, evidenciando una mejoría significativa de la disnea y la tos, con una saturación de oxígeno del 97 % a FiO2 ambiental. Actualmente, se mantiene estable y asintomática, acudiendo a sus controles mensuales en el consultorio externo, donde ha permanecido sin síntomas durante los últimos seis meses.
Discusión
La proteinosis alveolar pulmonar (PAP) es una enfermedad caracterizada por la acumulación anormal de lipoproteínas en los alveolos y macrófagos, resultante de un desbalance en la producción y eliminación del surfactante9. Este desbalance puede tener diferentes causas: primaria, vinculada a la alteración en la señalización del GM-CSF (factor estimulante de colonias de granulocitos y macrófagos), puede ser autoinmune (debido a la presencia de autoanticuerpos) o hereditaria (a través de mutaciones en los genes CSF2RB o CSF2RA, responsables de las subunidades del receptor de GM-CSF). También existe una forma secundaria asociada a enfermedades sistémicas o exposición a sustancias tóxicas inhaladas; así como una forma congénita provocada por mutaciones que afectan la producción o función del surfactante1,6.
El surfactante, compuesto por lípidos en un 90% y proteínas en un 10%, es fundamental para disminuir la reducir la tensión superficial en la zona de contacto entre aire y líquido, lo que previene el colapso alveolar y facilita la respiración. La síntesis y secreción del surfactante son funciones de las células epiteliales alveolares tipo II, mientras que los macrófagos alveolares se encargan de su degradación y eliminación10,11. La alteración en la señalización del GM-CSF juega un papel crucial en la patogénesis de la PAP2,5,9.
El diagnóstico de la PAP puede ser complejo y a menudo se confunde con una neumonía o asma, lo que conlleva a retrasos en el diagnóstico, que en algunos casos puede extenderse varios meses o incluso años3,12. En la PAP autoinmune, la presentación clínica suele ser insidiosa, con disnea progresiva, tos con esputo blanco espumoso, fatiga y pérdida de peso. La fiebre y la hemoptisis son menos comunes. En muchos casos, el examen físico puede resultar normal, aunque algunos pacientes pueden presentar crepitaciones pulmonares, dificultad respiratoria y cianosis5,6,13. En nuestro caso, la disnea y la tos fueron los síntomas principales, y la gasometría arterial mostró una ligera hipoxemia con una gradiente alveolo arterial (A-aDO2) elevada, hallazgos comunes en esta condición.
A pesar de que los marcadores inflamatorios y los exámenes de sangre, tales como la proteína C-reactiva (PCR), la velocidad de sedimentación eritrocitaria (VSG) y los niveles de lactato deshidrogenasa (LDH), pueden mostrar niveles elevados en pacientes con PAP, su utilidad diagnóstica sigue siendo limitada debido a su baja especificidad. Los anticuerpos circulantes contra GM-CSF son específicos de la PAP autoinmune y pueden ser indicativos, pero su presencia no es exclusiva de esta enfermedad, ya que también pueden encontrarse en individuos sanos1,6,12,13.
Los estudios de imágenes son fundamentales para el diagnóstico. En pacientes con PAP autoinmune, la radiografía torácica típicamente revela opacidades alveolares que son bilaterales y simétricas, con un aumento de la densidad en las áreas perihiliares. Aunque esta apariencia se asemeja al edema pulmonar, no se observan otros signos radiológicos de insuficiencia cardiaca, como cardiomegalia o derrame pleural5.
La tomografía computarizada de alta resolución (TCAR) del tórax puede revelar un patrón distintivo conocido como “crazy paving” o en empedrado, que se distingue por la presencia de áreas con atenuación en vidrio esmerilado, acompañadas de un engrosamiento de los septos inter e intralobulillares3,6. Es importante destacar que, aunque estos hallazgos sean típicos de PAP autoinmune, no son específicos y también pueden encontrarse en otras condiciones, como el edema pulmonar cardiogénico, la neumonitis por hipersensibilidad, la neumonía por Pneumocystis jirovecii y la carcinomatosis linfangítica5. Los hallazgos radiológicos en nuestro caso eran consistentes con el diagnóstico de PAP.
En la valoración de la función pulmonar de nuestro caso, se observó un patrón restrictivo leve, indicativo de un compromiso intersticial4,5,8,12. Es frecuente observar una disminución en la difusión del monóxido de carbono, que podría estar relacionada con la gravedad de la enfermedad. Cabe destacar que, en etapas iniciales, estos valores pueden encontrarse dentro de los rangos normales1.
Aunque los síntomas y los hallazgos radiológicos pueden ser sugestivos en casos típicos, la broncoscopia se considera comúnmente como el primer paso para confirmar el diagnostico de PAP. Se estima que aproximadamente el 75% de los casos sospechosos son confirmados mediante el lavado broncoalveolar (BAL)4,12,13. El BAL puede revelar un líquido de aspecto opaco y lechoso, característica de la enfermedad, y la citología puede mostrar macrófagos espumosos con gránulos eosinófilos y material amorfo que tiñe positivo en la tinción con ácido Schiff periódico (PAS) y negativo con azul alcián1,3. Sin embargo, estos hallazgos no permiten diferenciar el tipo de PAP.
La realización de una biopsia transbronquial puede aumentar el rendimiento diagnóstico especialmente cuando los estudios radiológicos y el aspecto macroscópico del BAL no son concluyentes3,5,13. La biopsia revela la presencia de un material eosinofílico denso en las vías respiratorias, con mínima inflamación intersticial4. En el caso de nuestra paciente, los hallazgos característicos de PAP se encontraron tanto en el estudio citológico de la muestra obtenida como en el estudio histopatológico de la biopsia transbronquial.
A pesar de los progresos en la comprensión de esta patología, las opciones terapéuticas siguen siendo limitadas y la decisión de empezar algún tratamiento se basa en la disminución de la función pulmonar y la presencia de dificultad respiratoria. Los objetivos terapéuticos son aliviar la sintomatología, optimizar la oxigenación y mejorar la calidad de vida. Pacientes con síntomas leves o sin ellos pueden ser manejados con cuidados de apoyo, seguidos de un monitoreo constante de la función pulmonar y radiografías de tórax4,9.
Si bien, el lavado pulmonar total (LPT), que implica la intubación selectiva y la instilación repetida de solución salina para eliminar el surfactante acumulado, es el tratamiento más utilizado y de primera línea para la PAP1,4, se han propuesto otras alternativas terapéuticas, como la inhalación con GM-CSF, el uso de rituximab, la plasmaféresis e incluso el trasplante pulmonar6,8.
El LPT se reserva para pacientes con disnea en reposo, una PaO2 en reposo inferior a 65 mmHg, un A-aDO2 en reposo mayor de 40 mm Hg o desaturaciones de oxígeno registrada durante la prueba de caminata de 6 minutos. En aproximadamente el 85% de los casos tratados, se observa una mejoría clínica y radiológica. Aunque no hay estudios exhaustivos sobre la eficacia ni la frecuencia óptima del procedimiento, algunas investigaciones sugieren que el LPT podría reducir la mortalidad a los 5 años en comparación con pacientes no tratados1,3,4,6. Dado los resultados de gasometría de nuestra paciente, no se consideró necesario iniciar este tratamiento durante su hospitalización.
La terapia de reemplazo de GM-CSF, que inicialmente administrada por vía subcutánea en pacientes con PAP autoinmune, ha sido generalmente bien tolerada. Sin embargo, su eficacia ha sido limitada en términos de alivio sintomático y mejora radiológica, especialmente en aquellos pacientes que no pueden someterse al LPT o que presentan PAP autoinmune refractario a este procedimiento. Aunque la mejora con esta terapia fue más lenta que con el LPT, evaluaciones posteriores de la terapia inhalada con GM-CSF mostraron una mejoría no solo en la disnea y la distancia recorrida durante la prueba de caminata de 6 minutos, sino también en la A-aDO2 y una mejora significativa en el intercambio gaseoso. Esto sugiere que la terapia inhalada con GM-CSF podría proporcionar más beneficios en la PAP autoinmune1,4,14-16.
Con respecto a la terapia de inmunomodulación, los corticosteroides sistémicos no ofrecieron beneficios en la PAP autoinmune e incluso podrían aumentar el riesgo de infecciones pulmonares. La plasmaféresis ha demostrado ser capaz de reducir con éxito los niveles circulantes de anti-GM-CSF en ciertos casos, potencialmente restaurando la hemostasia del surfactante. Sin embargo, la evidencia de la plasmaféresis sigue siendo limita y con resultados variables según los estudios publicados. Por otro lado, el rituximab, una agente anti-CD20, ha mostrado una mejora clínica significativa, particularmente en parámetros como la oxigenación arterial, la A-aDO2 y en las imágenes radiológicas después del tratamiento. A pesar de que no se ha registrado alteraciones en los niveles séricos de anti-GM-CSF, se notó una reducción de los niveles de IgG anti-MG- CSF en el BAL. Por lo tanto, aunque la evidencia disponible no respalda el uso rituximab como tratamiento de primera línea, tanto el rituximab como la plasmaféresis podrían considerarse como alternativas terapéuticas para la PAP autoinmune1,3,4,8,16.

