









 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
El Complejo de Esclerosis Tuberosa (CET) es una enfermedad neurocutánea de carácter hereditario, autosómico dominante causada por la mutación de dos genes: uno en el cromosoma 9 (9q34) que codifica la proteína hamartina y el otro en el cromosoma 16 (16p 13) que codifica la proteína tuberina, cuya función es regular e inhibir la producción de proteínas que activan la proliferación y diferenciación celular, por lo que el déficit de tuberina facilitaría el desarrollo de tumores además la hamartina interactúa con la tuberina formando un heterodímero que suprime uno de los controladores principales también de la proliferación y crecimiento celulares;1 forman un complejo regulador negativo de la vía receptora de insulina/fosfoinositido 3 cinasa / S6 cinasa, que inhibe la génesis tumoral.2 Sin embargo, del estudio clínico genético de pacientes con complejo de esclerosis tuberosa se ha visto que en hasta el 15 % de ellos no se han podido identificar las mutaciones antes mencionadas.3
Los tumores que se forman en esta enfermedad son semejantes a las células embrionarias, por lo que se cree que los efectos de la mutación se manifiestan muy tempranamente, los tumores que se desarrollan se llaman Hamartomas.1 Las manifestaciones neurológicas y cutáneas son variables, pudiendo ser las cutáneas las primeras en observarse en lactantes caracterizadas por áreas de hipomelanosis o nevos despigmentados hasta la formación de fibromas que aparecen generalmente al final de la infancia, la severidad en la presentación de dichas manifestaciones se relaciona al grado de mutacion.2
Los datos de incidencia se reportan de 1: 6000 personas, se afectan todas las razas, pero se considera poco frecuente en personas de raza negra además que podría existir una mayor incidencia en varones.2,3.
El complejo de esclerosis tuberosa CET tiene gran variabilidad en cuanto a su presentación clínica, y se considera con predisposición a enfermedad tumoral renal quística y sólida significativa.4
PRESENTACIÓN DEL CASO
Paciente varón de 24 años procedente de la ciudad de Tarija - Bolivia.
Antecedentes personales: Producto de segunda gestación de un embarazo pretérmino de 32 semanas con parto vaginal sin complicaciones, dentro de los antecedentes familiares abuelo paterno con diagnóstico de epilepsia, padre y madre sin antecedentes patológicos relevantes fallecidos, no se refiere la causa. Fue ingresado al servicio de medicina interna por presentar convulsiones tónico-clónicas generalizadas e intermitentes, con duración aproximada de 3 a 4 minutos cada crisis, acompañado de pérdida del conocimiento, el familiar acompañante relata historia de episodios convulsivos desde la niñez, además de presentar déficit neurocognitivo también desde edad temprana.
Al examen físico se observa pápulas eritematosas de diferentes tamaños distribuidas en el rostro (en las regiones de la frente, mentoniana, maseteriana, nasal y geniana) (Figura 1, A y B), en la parte endobucal se observó lesiones nodulares correspondiente a fibroma de mucosa gingival y lesiones del esmalte dental (Figura 2) a nivel de la encía superior; lesiones maculo-papulares hipocrómicas en región lumbar y fibromas periungueales (Figura 3).
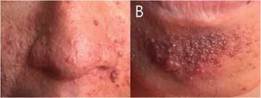
Figura 1: Vista frontal. Angiofibromas centrofaciales. A: Angiofibromas en región nasogeniana. B: Angiofibromas en región mentoniana.
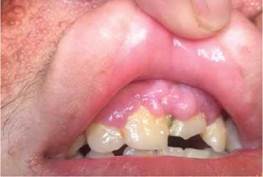
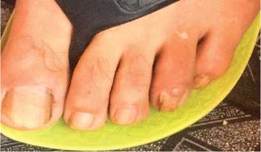
Exámenes complementarios
Ecografía abdominal: Reporta morfología renal alterada por aparentes masas en ambos riñones de apariencia quística.
La TAC de cráneo con contrasteC/C: reporta una asimetría ventricular, ventrículo lateral izquierdo dilatado, nódulos subependimarios y calcificaciones periventriculares.
La radiografía de tórax, electrocardiograma y ecocardiograma dentro de parámetros normales.
Diagnóstico: Se realizó en base a los criterios de la "U.S. NAtional Tuberous Sclerosis Associatión" considerándose un diagnóstico definitivo cuando están presente un criterio principal y dos secundarios, y el paciente presento angiofibromas faciales, fibromas ungueales, fibromas gingivales y quistes renales múltiples. VerTabla N° 1.
Tabla 1 Criterios Diagnósticos de CET según el Consenso Internacional de CET 2012
| TABLA N° 1 Criterios de diagnóstico actualizados para el complejo de esclerosis tuberosa 2012 |
|---|
| A. Criterios de diagnóstico genético. |
| La identificación de una mutación patogénica TSC1 o TSC2 en el ADN del tejido normal es suficiente para hacer un diagnóstico definitivo del complejo de esclerosis tuberosa. Una mutación patogénica se define como una mutación que inactiva claramente la función de las proteínas TSC1 o TSC2 (por ejemplo, mutación indeleble o sin sentido fuera del marco), impide la síntesis de proteínas (por ejemplo, deleción genómica grande), o es una mutación sin sentido. El efecto sobre la función proteica se ha establecido mediante la evaluación funcional (www.lovd.nl/TSC1, www.lovd /TSC2 y Hoogeveen-Westerveld et al., 2012 y 2013). Otras variantes de TSC1 o TSC2 cuyo efecto sobre la función es menos seguro no cumplen con estos criterios, y no son suficientes para hacer un diagnóstico definitivo de CET. Tenga en cuenta que del 10% al 25% de los pacientes con TSC no tienen una mutación identificada por las pruebas genéticas convencionales, y un resultado normal no excluye CET, ni tiene ningún efecto sobre el uso de criterios de diagnóstico clínico para diagnosticar CET. Dado que entre un 10 y un 25% de pacientes con CET tienen estudios mutacionales negativos, un resultado normal no excluye el diagnóstico y no afecta al uso de los criterios clínicos. |
| B. CRITERIOS DE DIAGNÓSTICO CLÍNICO. |
| CRITERIOS MAYORES |
| 1. Máculas hipomelanóticas (≥3, al menos 5 mm de diámetro) |
| 2. Angiofibromas (≥3) o placa cefálica fibrosa |
| 3. Fibromas unguales (≥2) |
| 4. Parche Shagreen |
| 5. Hamartomas retinianos múltiples |
| 6. Displasias corticales* |
| 7. Nódulos subependimarios |
| 8. Astrocitoma de células gigantes subependimarias |
| 9. Rabdomioma cardíaco. |
| 10. Linfangioleiomiomatosis (LAM)† |
| 11. Angiomiolipomas (≥2)† |
| CRITERIOS MENORES |
| 1. Lesiones cutáneas "confeti" |
| 2. Hoyos de esmalte dental (> 3) |
| 3. Fibromas intraorales (≥2) |
| 4. Parche acromático de la retina |
| 5. Quistes renales múltiples |
| 6. Hamartomas no renales |
| Diagnóstico Definitivo: Dos criterios mayores o un criterio mayor con ≥2 criterios menores. |
| Posible diagnóstico: Un criterio mayor o ≥2 criterios menores. |
* Incluye tubérculos y líneas de migración radial de la sustancia blanca cerebral.
† Una combinación de las dos características clínicas principales (LAM y angiomiolipomas) sin otras características no cumple con los criterios para un diagnóstico definitivo.
Nota: Tomado de Northrup H, Krueger DA. Tuberous Sclerosis Complex Diagnostic Criteria Update: Recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatric Neurology [Internet]. 2013 [citado 2020 mayo 01]; 49(4), 243-254. doi: 10.1016/j.pediatrneurol.2013.08.001
El tratamiento anticonvulsivo se inició en base a ácido valproico en dosis de 500 mg vía oral cada 12 horas; y haciendo uso también de midazolam 10 mg por vía endovenosa según requerimiento; sin embargo, durante su estancia hospitalaria presentó episodios convulsivos que no respondieron al tratamiento con posterior fallecimiento del paciente luego de permanecer 5 días internado.
DISCUSIÓN
El complejo de esclerosis tuberosa (CET) es una enfermedad poco frecuente, que en hasta dos tercios de los pacientes se debe a una mutación fresca, lo que sugiere una alta tasa de casos esporádicos al no haber el antecedente familiar. 1,2,5,6
Las manifestaciones clínicas son variadas, la mayoría de los órganos son susceptibles ya que la formación de hamartomas puede darse en múltiples sistemas, siendo más frecuentes las manifestaciones dermatológicas, renales y neurológicas, algunas manifestaciones neurológicas pueden ocurrir desde la infancia incluyendo espasmos infantiles, epilepsia de difícil tratamiento, discapacidades cognitivas y autismo, todas de expresión y gravedad variable.
La causa más común de la epilepsia son las malformaciones corticales focales. Los nódulos subependimarios son frecuentes y corresponden a hamartomas en el tejido subependimal de los ventrículos laterales que frecuentemente se calcifican. 7
Dentro de las manifestaciones dermatológicas; las máculas hipomelanoticas son prevalentes hasta en el 90% de niños menores de 5 años, pudiendo ser los primeros signos visibles de la enfermedad, además de los angiofibromas que también son comunes; la prevalencia aumenta con la edad y las lesiones progresan haciendo el componente fibroso más prominente. Los fibromas ungueales son menos comunes, generalmente aparecen en la adolescencia o en la edad adulta temprana, están ubicadas alrededor o debajo de las uñas; los parches de Shagreen son un criterio mayor dermatológico para el diagnóstico del complejo de esclerosis tuberosa y aparecen por lo general en la primera década de la vida inicialmente en la parte baja de la espalda; los fibromas gingivales son un criterio menor y se pueden incluir fibromas en cualquier sitio intraoral.1,2,8,9.
El paciente presento varios signos dermatológicos que coadyuvaron al diagnóstico, relacionándose con la edad que tenía, ya que varios signos progresan en tamaño o se hacen evidentes conforme los pacientes crecen, haciendo posible la sospecha diagnostica desde edades tempranas8,9.
La afectación renal suele ocurrir en el 80% de los casos incluyendo: angiomiolipoma, quistes renales y carcinoma de células renales en raras ocasiones10. Cuando se realizaron los estudios de imágenes se evidenciaron en el paciente lesiones quísticas renales, que en el momento no cursaba con insuficiencia renal como se reporta en otros casos publicados de pacientes adultos11.
El diagnóstico del complejo de esclerosis tuberosa (CET) es en base a Criterios diagnósticos, producto del consenso internacional de esclerosis tuberosa, la última actualización es del año 201212. VerTabla 1.
En base a ellos, el paciente presento como criterios mayores, angiofibromas faciales, fibromas ungueales, parche de Shagreen, nódulos subependimarios y como criterios menores los fibromas gingivales y los quistes renales, teniendo como resultado un diagnóstico definitivo del complejo de esclerosis tuberosa.
No existe un tratamiento específico, se ha estudiado el papel de varios grupos de medicamentos para la gran variedad de manifestaciones clínicas, por ejemplo, las lesiones cutáneas podrían ser candidatas a cirugía estética según criterio médico2,9, las convulsiones focales y generalizadas se tratan con fármacos anticonvulsivos , la elección del fármaco antiepiléptico debe seguir las resomendaciones de las guías de práctica clínica de epilepsia13. Las crisis epilépticas se pueden controlar con extirpación quirúrgica de las tuberosidades epileptógenas2.
Algo para tomar en cuenta es el papel de la vía receptora de insulina/fosfoinositido 3 cinasa / S6 cinasa que actúa como una vía de señalización y contribuidor crítico en el proceso de tumorogénesis14, un medicamento que actúa en este nivel es la rapamicina y en algunos estudios se ha demostrado que se asocia con la regresión de astrocitomas, existen otros medicamentos similares2.
La enfermedad renal quística suele responder a la descompresión quirúrgica, pero en caso de insuficiencia renal podría requerir diálisis o trasplante renal2,11.
CONCLUSIÓN
El CET es una enfermedad poco frecuente, que requiere un manejo multidisciplinario para tratar adecuadamente la progresión de la enfermedad, cuando se realiza el diagnóstico temprano se aumentan las posibilidades de
ofrecer una adecuada calidad de vida, es por ello que es importante contar con el conocimiento general para diagnosticar esta enfermedad en etapas tempranas, el diagnóstico es clínico y hay una gran variedad de signos clínicos sobre todo a nivel dermatológico que nos guían en el diagnóstico.