









 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO  uBio
uBio 
 Permalink
PermalinkRevista CON-CIENCIA
versão impressa ISSN 2310-0265
Rev.Cs.Farm. y Bioq vol.5 no.1 La Paz jun. 2017
ARTÍCULOS ORIGINALES
Establecimiento y evaluación de dos métodos de pre tratamiento de muestras de suelo para la extracción de ADN para el estudio de la diversidad bacteriana
Establishment and evaluation of two methods of pre-treatment of soil samples for the extraction of DNA for the study of bacterial diversity
DONAIRE FIGUEROA, KAREM1 PEREZ DUVAL, ANGELA1 ROMERO CALLE, DANITZA XIOMARA1 CÁRDENAS ALEGRÍA, OSCAR1 ÁLVAREZ ALIAGA, MARÍA TERESA1
1 Instituto de Investigaciones Fármaco Bioquímicas (IIFB), Facultad de Ciencias Farmacéuticas y Bioquímicas, UMSA.
FECHA DE RECEPCIÓN: 9 DE FEBRERO DE 2017 FECHA DE ACEPTACIÓN: 2 DE MAYO DE 2017
Resumen
La necesidad de ampliar los conocimientos respecto a los mecanismos bioquímicos y fisiológicos desarrollados por los microorganismos presentes en suelos requiere de una descripción completa de la diversidad microbiana, para lo cual en las últimas décadas se han desarrollado diferentes técnicas moleculares (qPCR, DGGE, T-RFLP, RAPD.) las mismas que requieren una adecuada técnica de extracción de ADN que aseguren el éxito de la descripción de la diversidad microbiana, considerando las características de las muestras de suelos a ser estudiadas.
Los protocolos de extracción de ADN generalmente utilizados están basados en la separación de los microorganismos de la matriz antes de la extracción de ADN mediante lisis física o química y por otro lado, la extracción directa del ADN microbiano a partir de muestras de suelo, sin embargo la presencia de sustancias húmicas y fenólicas afectan la calidad del ADN extraído, lo que repercuten en el desarrollo de posteriores estudios moleculares. La finalidad de este estudio fue la de establecer procedimientos de pre tratamientos de 3 tipos de muestras de suelo (arenoso, arcilloso y francos) para posteriormente describir la riqueza y diversidad bacteriana de las muestras en estudio mediante PCR DGGE. De esta manera se determinó que la adición de CaCO3 en muestras de suelos francos permite la identificación de una mayor diversidad y riqueza bacteriana (10 bandas). Asimismo, la adición de PVPP a suelos arenosos (8 bandas) y arcillosos (3 bandas) también permite obtener las características descritas anteriormente utilizando el método PCR-DGGE. Lo cual indica que los procedimientos de pre tratamiento con CaCO3 y PVPP son específicos para la extracción de ciertas comunidades microbianas.
PALABRAS CLAVE
Diversidad bacteriana, PCR DGGE, extracción de ADN, pre tratamiento de suelos.
Abstract
The knowledge about biochemical and physiological mechanisms by micro-organisms in soils are required for a complete description of microbial diversity, lately different molecular techniques have been developed to study this feature (qPCR, DGGE, T- RFLP, RAPD). DNA extraction techniques ensure the description of the microbial diversity success, according the soil samples characteristics.
Generally, DNA extraction protocols used for separation of microorganism of matrix soil before DNA extraction by physical and chemical lysis. Other protocol is direct extraction of microbial DNA from soil samples, humic acids and phenolic substances affect the quality of DNA, which affect the development of subsequent molecular studies.
The purpose of this study was to establish pretreatment procedures for different kind of soil samples (frank, sandy and clayey) in order to describe richness and bacterial diversity by PCR DGGE. In this sense, we determined the addition of CaCO3 in frank soils samples allows the identification of greater diversity and bacterial richness (10 bands) than the other method. Besides, PVPP pretreatment is no only useful to obtain bacterial diversity in sandy soil (8 bands), butalso in clayey soils (3 bands) soils by PCR-DGGE method. This indicates that the pretreatment procedures with CaCO3 and PVPP are specific for soil microbial community's isolation.
KEY WORDS
Bacterial diversity, PCR DGGE, DNA extraction, pretreatment of soils.
INTRODUCIÓN
Las comunidades microbianas desempeñan un rol crítico en procesos bio-geoquímicos, interactúan con las raíces de las plantas y también son constituyentes del suelo en la interface raíz-suelo. Los microorganismos promotores del crecimiento vegetal desempeñan un papel clave en la toma de nutrientes, siendo también esenciales para el desarrollo de otros organismos y el mantenimiento de la salud radicular, favoreciendo de esta manera la producción agrícola. Entre estos microorganismos, se encuentran las denominadas bacterias promotoras de crecimiento vegetal las mismas que son benéficas para el desarrollo de la planta, tolerancia a estrés ambiental, descomposición de la materia orgánica, la mineralización de nitrógeno, fósforo, azufre, produciendo además componentes bioactivos como vitaminas, hormonas y enzimas (Rashedul et al, 2012, Roose et al, 2001; Escalante et al, 2004, Coleman 2004, Rives et al, 2007 y Sagova, et al, 2008). Asimismo, estas bacterias benéficas, desintoxican el suelo de pesticidas, suprimen las enfermedades de plantas, inhibiendo el crecimiento de microorganismos patógenos del suelo, que son aquellos que pueden inmovilizar nutrientes, producir toxinas, sustancias pútridas que afectan el crecimiento y salud de las plantas. (Higa et al 1995). En este sentido se hace perentorio contar con una descripción más completa de la diversidad microbiana lo que posibilitaría ampliar nuestro conocimiento sobre los mecanismos bioquímicos y fisiológicos desarrollados por los microorganismos (Escalante et al, 2004).
Tradicionalmente los estudios para caracterizar la diversidad bacteriana en diferentes ambientes se basan en la suposición de que las técnicas de cultivo permiten recuperar la mayor parte de las comunidades bacterianas presentes en una muestra. Sin embargo, únicamente entre 0.1 y 10% de las bacterias en el ambiente son cultivables. No hay una explicación satisfactoria para entender la baja proporción de bacterias cultivables, y este fenómeno ha sido una seria limitación para estudiar la diversidad microbiana del suelo (Escalante et al., 2004).
Actualmente, existen herramientas o técnicas moleculares de la ingeniería genética o de la tecnología del ADN recombinante (Bolívar, 2004) que permiten la extracción, aislamiento, modificación y caracterización del ADN de cualquier organismo. Estas herramientas son fundamentales para comprender un número importante de procesos biológicos, estudio de la diversidad biológica en muestras ambientales (Boon et al., 2002; Lyautey et al., 2005; McIntosh et al., 2008), muestras de alimentos (Ampe et al., 1999; Ercolini, 2004) y además son técnicas útiles soportando el estudio de las comunidades microbianas en el suelo, favoreciendo de esta manera investigaciones agro biológicas que aportan al desarrollo de la agricultura dentro de un contexto multifactorial y multidisciplinario (Schneegurt et al, 2003; Escalante et al, 2004; Zaporozhenko et al, 2006). Dentro de estas técnicas podemos mencionar técnicas moleculares que han sido desarrolladas ampliamente como ser Reacción en cadena de polimerasa en tiempo real (qPCR), Microarray, electroforesisen gel con gradiente desnaturalizante (DGGE), y técnicas moleculares de huellas genéticas (T-RFLP, RAPD) (LaMontagne, et al, 2002).
Con la finalidad de describir la riqueza y diversidad microbiana de ecosistemas de suelos, numerosos procedimientos para el aislamiento y purificación de ADN han sido descritos (Sagova et al, 2008), debido a la complejidad relacionada a la heterogeneidad de las muestras. Se ha descrito que los diferentes tipos de muestras de suelos revelan una variación en cuanto al rendimiento del ADN, los factores que influyen al rendimiento son: 1) la diferencia entre métodos permite recuperar diferentes cantidades de ADN de diferentes especies, es así que el rendimiento es afectado por la especificidad del método, 2) la presencia de diferentes organismos a las bacterias y diferentes especies bacterianas, 3) finalmente el bajo rendimiento del ADN puede ser atribuido a la unión del ADN a ciertas partículas, tales como la arcilla y el humus estas partículas se encuentran cargadas negativamente, que facilita su unión a cationes de intercambio iónico; asimismo, sucede con la arcilla y la materia orgánica que pueden adsorber el material genético libre y/o degradarlo (Sagova et al, 2008). Lo que implica que la extracción de ADN a partir de la compleja matriz del suelo, representa una variedad de sustancias que pueden inhibir la actividad polimerasa, enzimas de restricción e incluso interferir con la hibridación.
Asimismo, la eficiencia de la extracción de ADN de los microorganismos presentes en el suelo depende del contenido celular, las características físico químicas pH, contenidos de materia orgánica (ácidos húmicos), contenido de arcilla, además agua, calcio entre otros, son factores importantes que influyen para obtener un protocolo de purificación y extracción eficaz (Sagova et al, 2008; Rhashedul et al, 2012). Para el aislamiento de ácidos nucleicos se recurre frecuentemente al uso de kits comerciales para la extracción de ADN de suelos y sedimentos como ser Favorgen, Power Soil DNA Isolation (Mobio), NorgenBiotek y UltraClean soil DNA (Hill et al.,2000; Martin et al., Diez et al., 2001, Castañeda et al, 2004; Tanase 2015).
Sin embargo, el rendimiento de la extracción de material genético con el empleo de estos kits puede ser bajo existiendo perdidas de comunidades microbianas durante el proceso de purificación, podría presentar una deficiente purificación proteica y de compuestos fenólicos que aun inhiben la amplificación del ADN y el costo de adquisición de estos kits. En consecuencia, existe la necesidad de adecuar y/o diseñar técnicas de extracción de ADN que aseguren el éxito de la PCR y posteriores estudios moleculares, considerando las características de las muestras (Robe et al., 2003; Pakpour, et al., 2013) y que las mismas sean apropiadas para el estudio de ecosistemas complejos tales como los suelos.
Los protocolos de extracción de material genético a partir de muestras de suelo pueden ser clasificados en dos grupos: 1) Separación de los microorganismos de la matriz antes de la extracción de ADN mediante lisis física o química y, 2) Extracción directa del ADN microbiano a partir de muestras de suelo.
El presente trabajo tiene por finalidad es optimizar la extracción directa de material genético de microrganismos a partir de 3 tipos de muestras de suelo (arenoso, arcilloso y francos), incluyendo dos procesos de pre-tratamiento de las muestras consistentes en la adición de carbonato de calcio y de PVPP (polivinilpolipirrolidona), para posteriormente describir la riqueza y diversidad bacteriana de las muestras en estudio mediante PCR DGGE.
MATERIALES Y MÉTODOS
Colección de muestras de suelos
Se colectaron tres tipos de muestras de suelos en tubos Falcón de 50 ml: i) suelo francos, recolectado de un barranco en la zona Santa Rosa Grande (La Paz), ii) suelo arenoso, recolectado de la zona Villa Fátima (La Paz) y iii) suelo arcilloso, mismo que fue adquirido de una librería Paty. Posteriormente, las muestras fueron homogenizadas, tamizadas y secadas en mufla a 50 °C durante 24 horas.
Extracción y purificación de material genético a partir de muestras de suelo
Las muestras de suelo fueron sometidas a dos tipos de protocolos de extracción y purificación de ADN.
Extracción y purificación de material genético a través del Kit comercial Favorgen
Las extracciones de material genético de las muestras de suelos fueron procesadas utilizando el protocolo establecido por el Kit Favorgen por triplicado. Se adicionó 0,5 g de muestra de suelo en cada tubo ep-penddorf que contenía 200 mg de perlas de vidrio y 600 ul de tampón de SDE1, las mezclas fueron homogenizadas durante 5 min y fueron incubadas a 70 °C durante 10 minutos. Durante la incubación las suspensiones fueron homogenizadas cada 5 min., posteriormente, los tubos eppendorf se centrifugaron a 14 000 rpm durante 5 min, se añadió 200 ul de tampón SDE2 a las suspensiones y se mantuvieron las muestras en baño de hielo durante 5 min. Consecutivamente, los tubos fueron centrifugados a 14 000 rpm durante 5 min, los sobrenadantes fueron transferidos a otros tubos eppendorf 1,5 ml, se adicionaron 1 volumen de isopropanol y centrifugaron a 14 000 rpm durante 10 min. Se eliminó cuidadosamente los sobrenadantes, para lo cual se invirtieron los tubos sobre papel toalla durante 1 min y se añadió 200 ul de tampón de elución precalentado. Subsecuentemente, a las suspensiones se adicionó 100 ul de tampón SDE3, se homogenizó e incubó a temperatura ambiente durante 3 min y se centrifugó a 14 000 rpm durante 2 min. Los sobrenadantes fueron transferidos a otros tubos eppenddorf y se adicionaron 1 volumen de buffer SDE4 y 250 ul de etanol al 70 %. Las suspensiones obtenidas fueron colocadas en columnas SDE, se centrifugaron por 1 min y la soluciones eluídas fueron eliminadas. Las columnas SDE fueron transferidas a otros tubos eppendorf, se añadió 750 ul de tampón de lavado a las columnas SDE y se centrifugaron a 14 000 rpm por 1 min, se descartó las soluciones eluídas, este procedimiento se realizó tres veces. Consecutivamente, los tubos fueron centrifugados a 14000 rpm durante 3 min. Se trasvasó las columnas SDE en tubos eppenddorf, se añadió centro de la membrana de la columna de SDE 200 ul de tampón de elución precalentado. Finalmente, las columnas SDE fueron incubadas a temperatura ambiente por 2 min y fueron centrifugadas a 14 000 rpm.
Extracción y purificación de material genético a través de lisis celular directa sin pre-tratamiento
Se pesaron 0,5 g de cada muestra de suelo en tubos eppendorf, seguidamente se adicionaron 700 uL de buffer de lisis [100 mM de Tris-HCl, 100 mM de EDTA pH=8,100 mM de fosfato de sodio di hidratado, 1.5 % hexadecyl methyl ammonium bromide (CTAB) pH=8] y 50 uL de proteinasa K(10 mgmL-1) (Fatimaetal., 2011). Las suspensiones fueron sometidas a agitación constante a 225 rpm por 45 min a 37 °C, transcurrido ese tiempo se adicionó 150 uL de SDS al 20% y 50 uL de lisozima (10 mg mL-1), las suspensiones fueron nuevamente sometidas a agitación a 750 rpm durante 2 horas a 65 °C y se centrifugó a 8500 rpm por 10 min. Los sobrenadantes fueron trasvasados a tubos eppendorf nuevos y al pellet se adicionó 200 uL de buffer de lisis con 30 uL de SDS al 20% y se incubó a 750 rpm por 15 min a 65 °C. Posteriormente, los sobrenadante fueron trasvasados a otros tubos eppendorf y el pellet fue eliminado. A las suspensiones colectadas se adicionaron 1 volumen de cloroformo: alcohol-isoamílico (24:1) incubándose a 20 ° C durante 20 minutos y consecutivamente se centrifugaron a 10 000 rpm por 10 min. Los sobrenadantes fueron trasvasados a otros tubos eppendorf y se adicionó 1 volumen de isopropanol a cada uno de los tubos y se mantuvo en baño de hielo por 20 min. Nuevamente se centrifugaron los tubos a 14 000 rpm por 15 minutos y se eliminaron los sobrenadantes. Consecutivamente, a los pellets se adicionaron 200 ul de etanol al 70%, se centrifugaron los tubos a 14 000 rpm por 3 min, los sobrenadantes fueron eliminados y el ADN obtenido de cada tubo eppendorf fue secado a 55 °C. Finalmente se adicionaron 200 ul de buffer TE y los tubos fueron conservaron a -20 °C hasta su utilización.
Purificación de ADN
Para realizar la purificación de ADN se utilizaron columnas que contenían papel filtro Whatman 0.5, algodón y Sephadex G100.
a. Preparación de las columnas: se utilizaron columnas de 1.25 cm de diámetro (jeringa de 5ml), se colocó papel filtro Whatman 0.5 en el interior de la columna junto con algodón y 1.5 mL Sephadex G 100 (suspensión de 2.1 g/10 mL de buffer TE).
b. Elución del ADN: se realizaron 3 lavados de las columnas con buffer TE a través de la presión ejercida con el embolo de la jeringa, se adicionó 200 ul del ADN extraído a cada columna. El ADN eluido fue colectado en un tubo eppendorf. Además, descartando la suspensión de Sephadex G 100 de las columnas, tanto el algodón como el papel filtro fueron colocados en un vial que contenía 3 ml de buffer TE, se incubó durante 15 min y posteriormente el contenido de los viales fue transvasado a tubos eppendorf. En ambos, en la solución de ADN eluido y en la solución ADN recuperado mediante el lavado con buffer TE, se añadió un volumen de isopropanol, se mantuvo en baño de hielo por 20 min, posteriormente los tubos fueron centrifugados a 14 000 rpm por 15 minutos, se eliminó el sobrenadante y se adicionó 200 ul de etanol al 70%, se centrifugó a 14 000 rpm por 3 min, y se eliminó el sobrenadante. El ADN obtenido fue deshidratado a 55 ° C, finalmente se adicionó 200 ul de buffer TE y conservó a -20 °C.
- Extracción y purificación de material genético a través de lisis celular directa con pre-tratamientos
a) Pre- tratamiento con carbonato de calcio
Se pesaron 0.5 g de cada muestra de suelo en tubos eppendorf y se adicionaron 500 uL de CaCO3 1M y se incubaron a temperatura ambiente durante 1h, posteriormente los tubos eppendorf fueron centrifugados a 8500 rpm por 10 min y los sobrenadantes fueron eliminados (Cermak et al, 2008).
b) Pre- tratamiento de PVPP
Se pesaron 0.5 g de cada muestra de suelo en tubos eppendorf y se adicionó 1 volumen de polivinilpolipirrolidona (PVPP) al 1%, los tubos fueron sometidos a agitación constante en shaker orbital a 100 rpm por 15 min y finalmente se eliminó el sobrenadante.
Consecutivamente a ambos pre- tratamientos, se estableció el procedimiento de extracción y purificación modificado de acuerdo a los autores Sharma et al., 2007 y Wang et al., 2008.
Amplificación de la región variable V3 del 16S ARNr
A partir de las distintas muestras extraídas y purificadas de ADN se amplificaron por PCR fragmentos del gen codificante región variable V3 del 16S ARNr. Se utilizaron primers denominados 341F-GC clamp (5-CCT ACG GGA GGC AGC AG CGC CCGCCGCGC CCC GCGCCC GTC CCGCCGCCC CCGCCC-30 y 534R (5'-ATT ACC GCG GCT GCT GG-3') que amplifican una región de 234pb. Se utilizó 20 uL de mix para la amplificación, [0.03 UuL-1 de Taq polimerasa, 1x de buffer, 0.8 uM de cada primer, 0.2 mM de dNTPs, 1.5 mM de MgCl2, 0.5 mg mL-1 de albúmina de suero bovino (BSA)] y 5 ug mL-1 de ADN. Las condiciones de amplificación del ADN por PCR fueron: Desnaturalización inicial a 94 °C por 10 min, seguida de 15 ciclos de Desnaturalización a 94°C por 1 min, Alineamiento a X°C por 1 min, (X representa la temperatura de alineamiento en la cual se disminuye 2 °C desde 66 °C hasta 52°C, la disminución de la temperatura de alineamiento se estableció cada 3 min) y Elongación de la cadena a 72°C por 3 min. Consecutivamente, nuevamente 15 ciclos de Desnaturalización a 94 °C por 1 min, Alineamiento a 55 °C por 1 min y Elongación de la cadena a 72 °C por 3 min y finalmente, la Elongación final a 72 °C por 10 min. Para cada reacción se utilizó un control negativo equivalente a un volumen de agua destilada estéril (Wang et al, 2008). Posteriormente se realizó la corrida electroforética en geles de agarosa al 1.5%.
Condiciones de la electroforesis en gel con gradiente desnaturalizante (DGGE)
Para separar los distintos fragmentos amplificados de la región variable V3 del 16S ARNr, se realizó una electroforesis en gel con gradiente desnaturalizante (DGGE) utilizando un gradiente de urea-formamida siguiendo las indicaciones descritas por Muyzer et al.,1998.
Se prepararon geles de poliacrilamida al 8 % (v/v) con diferentes gradientes desnaturalizantes de urea-formamida de 40 a 60%, (100% de desnaturalizante definido como: 7 M de urea y 40% de formamida desnaturalizante), acrilamida bis acrilamida (37.5:1) al 8%. En cada pozo del gel se sembraron 10 ul del producto de amplificación y la electroforesis se realizó a 60 V durante 7 horas en bufferTAE 1x colocado en el tanque de electroforesis a 60 °C (As-cher et al., 2004). Los geles de poliacrilamida fueron revelados utilizando la tinción de nitrato de plata (Sánchez, 2015).
Análisis estadístico
Se realizó correlación de Spearman para determinar la relación entre el tipo de suelo y procedimiento de extracción, análisis Kruskal-Wallis (p<0,05), para establecer las diferencias entre el rendimiento, índices de pureza, tipo de muestras y procedimiento de extracción de ADN, análisis Kruskal-Wallis (p<0,05) para identificar la diferencia entre el número de bandas obtenidas mediante el PCR-DGGE y procedimientos de extracción empleados, se realizó análisis de clúster método UPGMA para determinar la similitud en la composición de las comunidades entre los distintos tipos de suelo. Estos análisis se realizaron utilizando el software R Project.
RESULTADOS Y DISCUSIÓN
La eficiencia de la extracción y purificación depende de las características físicas químicas de la muestra, la elección del protocolo debe estar relacionada al rendimiento y la calidad del material genético obtenido, además este debe ser representativo de toda la comunidad microbiana presente en el nicho ecológico en estudio. La extracción del material genético dependerá del tipo de muestra de suelo (Niemi et al, 2001; Schneegurt et al, 2001; Robe et al, 2003; Young et al, 2014).
Determinación de la calidad y rendimiento del material genético post Extracción y Purificación.
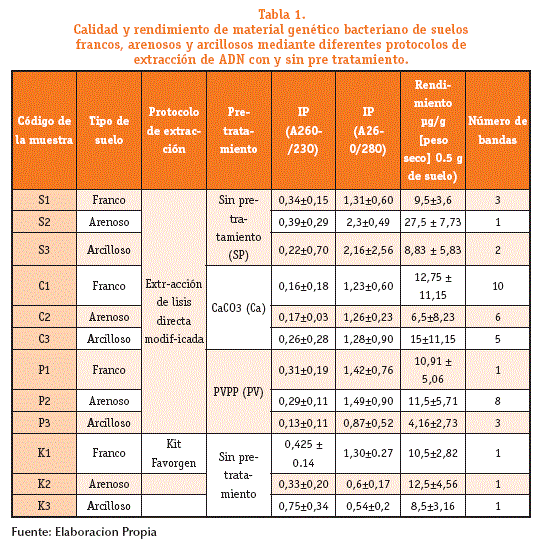
En el presente estudio inicialmente se desarrollaron dos protocolos de extracción, en el primer protocolo se realizó utilizando un método modificado y otro mediante el Kit Favorgen. Mediante el DGGE se determinó una sola banda utilizando el Kit Favorgen y mediante el protocolo de extracción modificado se obtuvo hasta tres bandas en muestras de suelos francos (Figura 1 y tabla 1). Es así, que se adicionó dos procedimientos a la extracción de material genético modificado; a) pre tratamiento con carbonato de calcio y b) pre tratamiento con PVVP para obtener un mayor número de bandas, es decir determinar mayor cantidad de géneros o especies bacterianas. Utilizando el pre tratamiento con carbonato de calcio se obtuvo hasta diez bandas en muestras de suelo arenosos.
- Obtención de material genético por el uso del Kit comercial Favorgen y a través de lisis celular directa sin pre-tratamiento
Para evaluar la pureza del material genético obtenido de las muetras de suelo, se determinó los indices de pureza (IP), según las siguientes relaciones de absorvancias: A260/230 (Ácidos nucleicos/Ácidos húmicos) y A260/280 (Ácidos nucleicos /proteína). Si el IP A260/230 es superior a 2 indica una pureza ideal de ADN, valores inferiores representa contaminación por ácidos húmicos. La relación A260/280 mayor a 1.8, implica un ADN puro libre de proteínas (Liesacketal, 1997; Bürgmann, etal; 2001; Fátima et al, 2011). En este sentido, el procedimiento propuesto para la extracción de ADN bacteriano a partir de las muestras en estudio, no remueven completamente los acidos húmicos y/o compuestos fenolicos incluyendo el kit comercial. Por otro lado, la contaminación por proteinas en la mayoría de los casos fue inferior a 1.8 a excepción excepcion de las muestras S2 y S3, como se observa en la tabla 1.
Además, no se identificó diferencias significativas entre el tipo de muestra, tipo de procedimiento, IP de A260/230, IP de A260/280 y rendimiento de ADN (Kruskal Wallis, p>0,05). Sin embargo, el procedimiento empleado de acuerdo el pre tratamiento dependera de la natureleza de la muestra o tipo de suelo empleado.
Con respecto al rendimiento de ADN, el mayor obtenido fue de 27.7 ug/g (S2) y el menor rendimiento fue de 4.16 ug/g en (P3), las muestras pre tratadas con carbonato de calcio a excepción de suelos arenosos, mostró los mejores rendimientos en suelos francos y arcillosos, el análisis ha sido calculado los valores de la media por triplicados (figura 1 y tabla 1).
En este trabajo se utilizó la lisis directa, debido a que es más rápido, se obtiene mayor rendimiento de ADN y es representativo de la comunidad microbiana presente en la muestra. Sin embargo, existen desventajas de este método como ser las extracciones adicionales de componentes orgánicos tales como ácidos húmicos, compuestos fenólicos, aromáticos entre otros que interfieren en los subsecuentes análisis (Amsaleg et al, 2001; Schneegurt et al, 2003).
Po otro lado, se ha descrito que el pre tratamiento de las muestras de suelo con CaCO3, el cual es eficiente para la obtención de ADN (Cermak et al, 2008; Sagova et al, 2008; Wang et al, 2008). Por otro lado, el PVPP puede remover los ácidos húmicos, aunque se ha descrito una pérdida de material genético en la extracción. Este es ineficaz durante la lisis celular, pero es útil en columnas de purificación del material genético para eliminar compuestos fenólicos u otras sustancias orgánicas (Steffan et al, 1988; Robe et al 2003; Cáceres et al, 2012; Singh et al, 2013), en el presente trabajo se observó que el rendimiento del material genético de las muestras pre tratadas es similar al kit comercial (figura 1).
Amplificación de la región variable V3 del 16S ADNr
Los diferentes extracciones y pre tratamientos aplicados en las diferentes muestras se valoraron mediante la capacidad de disminuir los inhibidores presentes, mediante la amplificación de un segmento de la región variable (V3) del gen 16S ADNr (Wang et al, 2008), en el presente trabajo se utilizó el mismo método de purificación, los extractos de ADN obtenidos presentaron alta cantidad de contaminación proteica y ácidos húmicos, que no afectaron a la PCR, asimismo se utilizó como coadyuvante BSA para mejorar la eficiencia de la amplificación en todas las muestras, a 0,8 ug uL-1 incrementa la eficiencia de la PCR actuando como una proteína captadora de iones que pueden ser inhibidores de la Taq polimerasa (La Montagne et al, 2002).
Condiciones de desarrollo de electroforesis en gel con gradiente desnaturalizante (DGGE)
En la figura 1 se observa el fragmento amplifcado de la region V3 del gen 16S ADNr en geles de poliacrilamida con en gradiente desnaturalizante, de acuerdo al número de bandas, se puede verificar la diversidad microbiana presente (Agnellia et al, 2004), en los tres tipos de muestras de suelo utilizados en este estudio el mayor bandeaje obtenido se encuentran en muestras procedentes de suelo francos (figura 2b carriles 4 y 5), arenoso y arcilloso (figura 2b carriles 10 y 11), las cuales corresponden aquellas muestras que fueron sometidas al pre tratamiento con CaCO3 y con PVPP respectivamente en contraste a la muestras sin pre tratamiento solamente presentaron una banda. Asi mismo, se observa que el uso de carbonato es mas eficiente cuando se utiliza muestras procedentes de suelos francos, no así en muestras arenosas y arcillosas (figura 2b carriles 6 y 7), las muestras tratadas con PVPP presentaron el mayor numero de bandas en muestras arenosas y arcillosas (figura 2b carriles 10 y 11), se debe utilizar el pre tratamiento cuando se realiza un método directo de extracción de material genético, además de considerar según el tipo de muestra el uso de CaCO3 y/o PVPP.
En las muestras que presentaron mayor numero de bandas se observaron bandas consenso, representaria la presencia de comunidades bacterianas frecuentes en las diferentes muestras analizadas, con respecto al análisis realizado con el kit comercial se observa solo una banda consenso (figura 1 a carriles 1 al 3).

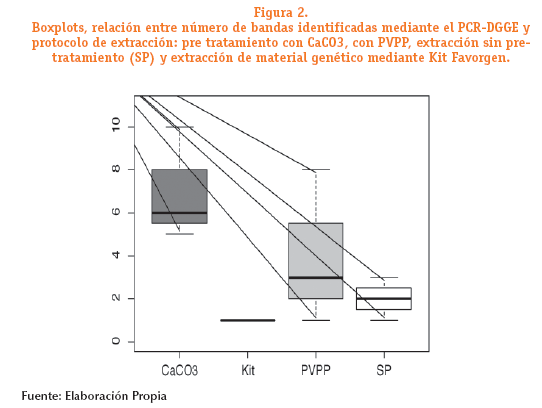
Asimismo, se determinó que no existen diferencias estadísticamente significativas entre el número de bandas y protocolo de extracción (Kluskal Wallis p value>0,05). No obstante, el procedimiento de extracción mediante el cual se identificó mayor número de bandas fue mediante el pre-tratamiento con CaCO3 y el protocolo que identifico menor número de bandas fue mediante el Kit Favorgen. La mediana del número de bandas es 6 mediante el pre-tratamiento con CaCO3, con un valor máximo de hasta 10 bandas mediante este protocolo, por otro lado, mediante el kit de extracción el valor de la mediana fue de 1 (figura 2).
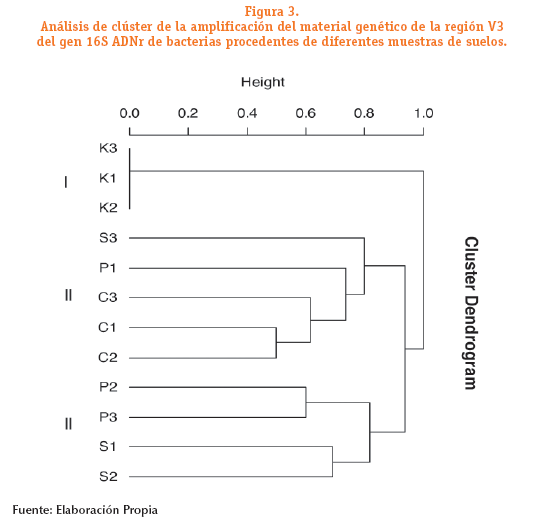
De acuerdo al análisis de clúster realizado se identificó la estructura de las comunidades bacterianas representada por la formación de tres clados o agrupamientos: clado I constituido por K1, K2 y K3, en el cual se identificó la misma comunidad bacteriana en los tres tipos de suelo, el clado II representado por S1, P1 C1, C2 y C3, en el cual las comunidades bacterianas en S1 están presentes en P1, C1, C2 y C3 y el clado III, conformado por P2, P3, S1 y S2, donde se observa que las comunidades bacterianas se agrupan en relación al pre tratamiento empleado. Los procedimientos de pre tratamiento de extracción de ADN, son específicos para los distintos tipos de comunidades bacterianas y del tipo de suelo.
Como señala Escalante et al, 2004, es posible caracterizar la diversidad no cultivable presente en diferentes ambientes por medio del análisis del meta-genoma bacteriano. La información generada permitirá comprender la función de las comunidades bacterianas en los ciclos biogeoquímicos que mantienen a la biosfera, y cómo la actividad humana local y global puede alterar la diversidad microbiana. El análisis de la diversidad genética y metabólica del metagenoma bacteriano de muestras de suelo permite extraer y explotar su diversidad metabólica, incluyendo la de aquellos microorganismos considerados cultivables, y de aquellos aún no descubiertos. Esta metodología permitirá identificar actividades bacterianas novedosas con aplicaciones biotecnológicas potenciales.
CONCLUSIONES
La extracción de ADN mediante los procedimientos de lisis directa, purificación con Sephadex G 100 y el kit Favorgen sin pre tratamiento de las muestras de suelos francos, arenosos y arcillosos, no fue óptima respecto al estudio de la diversidad y riqueza bacteriana. Sin embargo, la aplicación de un pre tratamiento consistente en la adición de CaCO3 en muestras de suelos francos permitió la identificación de una mayor diversidad y riqueza bacteriana. Asimismo, la adición de PVPP a suelos arenosos y arcillosos también permitió obtener las características descritas anteriormente utilizando el método PCR-DGGE, lo cual indica que los procedimientos de pre tratamiento con CaCO3 y PVPP son específicos para la extracción de ciertas comunidades bacterianas y del tipo de suelo.
Los resultados de este estudio señalan que el protocolo de extracción con la inclusión de los pre tratamientos descritos, representa una metodología útil y accesible para los estudios ecológicos bacterianos de suelos francos, arenosos y arcillosos con la facilidad de obtener la estructura y diversidad de las comunidades bacterianas.
AGRADECIMIENTOS
Este trabajo ha sido realizado gracias al apoyo académico del Dr. Rolando Sánchez Montaño, la financiación del proyecto "Análisis tóxico-genético del agua de afluentes que abastecen la planta de tratamiento de Achachicala provenientes de las lagunas Milluni Chico, Grande y Janko Kkota; su incidencia en la salud humana y la adaptación de la flora microbiana.
REFERENCIAS BIBLIOGRÁFICAS
Ascher, J., Corti, G & Ceccherini. (2004). Distribution of microbial communities in a forestsoil profile investigated by microbial biomass, soil respiration and DGGE of total and extracellular ADN. Soil Biology & Biochemistry, 36, 859-868. [ Links ]
Agnellia, A., Ascherb, J., Cortia, G., Ceccherinib, M. T., Nannipierib, P. & Pietramellara. (2004). Distribution of microbial communities in a forest soil profile in-vestigated by microbial biomass, soil respiration and DGGE of total and extracellular ADN. Soil Biology & Biochemistry, 36, 859-868. [ Links ]
Ampe, F., Ben, O.N., Moizan, C., Wacher, C., Guy, JP. 1999. Polyphasic study of the spatial distribution of microorganism-sin mexican pozol, a fermented mai-ze dough, demonstrates the need for cultivationindependent methods to investigate traditional fermentations. Applied and Environmental Microbiology. 65:5464-5473. [ Links ]
Amsaleg R., Sillam G., Harry M., (2001). Extraction and purification of microbial DNA from soil and sediment samples. Soil Ecology 18,47-60. [ Links ]
Coleman, CD., Crossley, DA. and Hendrix, PF. (2004). Historical Overview of Soils and the Fitness of the Soil Environment. En Kelly Sonnack (coord.), Fundamentals of Soil Ecology (pp. 57-61). San Diego, California: Copyright, Elsevier Inc. [ Links ]
Boon, N., De Windt, W., Verstraete, W., Top, E.M. 2002. Evaluation of nested PCR-DGGE (denaturing gradient gel electrophoresis) with group-specific 16S rDNA primers for the analysis of bacterial communities from different was-tewater treatment plants. FEMS Micro-biology Ecology. 39:101-112. [ Links ]
Bolívar, F. 2004. Ingeniería genética: las herramientas moleculares y los métodos para aislar, caracterizar y manipular el DNA. In: Fundamentos y Casos Exitosos de la Biotecnología Moderna. Bolívar, F. (ed). El Colegio Nacional, México. pp: 57-84. [ Links ]
Braid, M., Daniels, L & Kitts, C. (2003). Remo-val of PCR inhibitors from soil ADN by chemical flocculation. Journal of Micro-biological Methods, 52, 389-393. [ Links ]
Bürgmann, H., Pesaro, M., Widmer, F. & Ze-yer, J. (2001). A strategy for optimizing quality and quantity of ADN extracted from soil. Journal of Microbiological Methods, 45, 7-20. [ Links ]
Cáceres, P., Cordero, C., González, G., Qui-roz, K., Bobadilla, J.C., Bravo, C., Cali-gari, P. D., Carrasco, B. & García, R. G. (2012). Efficient protocols for the ex-traction of microbial ADN from the rhi-zosphere of hydrophilic forests in Chile. Cien. Inv. Agr, 39(3),585-592. [ Links ]
Castañeda, A., McEwen, J., Hidalgo, M. & Castañeda, E. (2004). Evaluación de varias técnicas de extracción de ADN de Cryptococcus spp. a partir de muestras ambientales. Biomédica, 24, 324-31 [ Links ]
Cermak, L., Novatna, J. & Pihackova, K. (2008). Invovative Methodos for Soil ADN Purification Tested in Soil With Wi-dely Differing Characteristics. Applied and Environmental Microbiology, 74, 2902-2907. [ Links ]
Díez D., Alió C., Marsh T., Massana R., (2001). Application of Denaturing Gradient Gel Electrophoresis (DGGE) To Study the Diversity of Marine Picoeu-karyotic Assemblages and Comparison of DGGE with Other Molecular Techni-ques, 67,942-945. [ Links ]
Escalante, A. L., Gosset, G.L., Martínez, M.A & Bolívar, F.Z. (2004). Diversidad bacteriana del suelo: Métodos de estudio no dependientes del cultivo microbiano e implicaciones biotecnológicas. Agro-ciencia, 38 (6), 583-592. http://dx.doi.org/10.4236/ajmb.2013.34028 Published Online October 2013. [ Links ]
Ercolini, D. 2004. PCR-DGGE fingerprinting: Novel strategies for detection of micro-bes in food. Journal of Microbiological Methods. 56:297314. [ Links ]
Fatima, F., Chaudhary, I., Ali, J., Rasgoti, S, & Pathak, N. (2011). Microbial ADN extrac-tion from soil by different methods and its PCR amplification. Biochem. Cell. Arch, 11.1-6. [ Links ]
González-de la Cruz, U., González, D., de la Cruz, M., Rojas-Herrera, R. & Zamudio-Maya3. (2011). Protocolo para la extracción de ADN metagenómico bacteriano del langostino Macrobrachium carcinus. Tropical and Subtropical Agroecosys-tems, 14, 875-883 875. [ Links ]
HMI G.T., Mitkowski N.A., Aldrich-Wolfe L., Emele L.R., Jurkonie D.D., Ficke A., Mal-donado-Ramireza S.,Lyncha S.T., Nelso-na E.B.(2000). Methods for assessing the composition and diversity of soil micro-bial communities,15,25-36. [ Links ]
Higa, T. (1995). Effective microorganisms: Their role in Kyusei Nature Farming and sustainable agriculture. In J.F. Parr, S.B. Hornick, and M.E. Simpson (ed.) Pro-ceedings of theThird International Con-ference on Kyusei Nature Farming. U.S. Department of Agriculture, Washington, D.C., USA. (In Press). [ Links ]
LaMontagne, L. G., Michel, F. C., Holden, P.A. & Reddy, C.R. (2002). Evaluation of extraction and purification methods for obtaining PCR-amplifiable ADN from compost for microbial communi-ty analysis. Journal of Microbiological Methods, 49, 255-264. [ Links ]
Liesack, W.P., Jansen, H., Rainey, F. A., Ward, R. N. & Stackebrandt, E. (1997). Microbial diversity in soil: the need for a com-bined approach using molecular and cultivation techniques, Modern Micro-biology, 375-439. [ Links ]
Lyautey, E., Lacoste, B., Ten-Hage, L., Rols, J-L., Garabetian, F. 2005. Analysis of bacterial diversity in river biofilms using 16S rDNA PCR-DGGE: methodologicalset-tings and fingerprints interpretation. Water Research. 39:380-388. [ Links ]
Martin-Laurent,F., Philippot,L., Hallet s., Chaussod R., Germon J., Soulas G., Ca-troux G. (2001). DNA Extraction from Soils: Old Bias for New Microbial Diversity Analysis Methods, 67,354-359. [ Links ]
McIntosh, D., Ji, B., Forward, B.F., Puvanen-dran, V., Boyce, D., Ritchie, R. 2008. Cul-tureindependent characterization of the bacterial populations associated with cod (Gadus morhua L.) and live feed at an experimental hatchery facility using denaturing gradient gel electrophoresis. Aquaculture. 275:42-50. [ Links ]
Muyzer, G & Smalla, K. (1998). Application of desnaturing gradient gel electrophoresis (DGGE) and temperature gradient gel electrophoresis (TGGE) in microbial ecology. Antonie van Leeuwenhoek, 73, 127-141. [ Links ]
Niemi, R., Heiskanen, I., Wallenius, K., Linds-trom, K. (2001). Extraction and purification of DNA in rhizosphere soil sam-ples for PCR-DGGE analysis of bacterial consortia. Journal of Microbiological Methods, 45, 155-165. [ Links ]
Pakpour, S., Olishevska, S.V., Prasher, S.O., Milani, A.S., & Chénier, M. R. (2013). ADN extraction method selection for agricultural soil using TOPSIS multiple criteria decision-making model. American Journal of Molecular Biology, 3, 215-228. [ Links ]
Sánchez, RM. (2015). Guía de Trabajos Prácticos de Biología Molecular. 27- 29, 70. [ Links ]
Sagova, M., Cermak, L., Novotna, J., Plhac-kova, K., Forstova, J & Kopecky, J. (2008). Innovative Methods for Soil ADN Purification Tested in Soils with Widely Differing Characteristics. Applied and Environmental Microbiology, 74 (9), 2902-2907. [ Links ]
Singh, R., Sivasubramani, K., Jayalakhsmi, S., (2013). Simple, rapid method for direct isolation of metagenomic dna from pi-chavaram mangrove sediment,2277-3827. [ Links ]
Steffan, R.J., Goksøyr, J., Bej, A.K., Atlas, R.M., 1988. Recovery of DNA from soils and sediments. Appl. Environ. Microbiol. 54, 2908-2915. [ Links ]
Roose, C.L., Garnier, E. & Harry, M. (2001). Extraction and purification of microbial ADN from soil and sediment samples. Applied Soil Ecology, 18, 47-60. [ Links ]
Schneegurt, M.A., Dore, S. Y. & Kulpa, Ch. J. (2003). Direct Extraction of ADN from Soils for Studies in Microbial Ecology. Curr. Issues Mol. Biol, 5,1-8. [ Links ]
Sharma, P. K., Capalash, N. & Kaur, J. (2007). An improved method for single step purification of metagenomics ADN. Mol Biotechnol. http://dx.DOI10.1007/s12033-007-0015-3,1-3 [ Links ]
Rashedul, M.D., Sultana, T., Melvin, M.J., Cheon, J. & Sa, J. (2012). Comparisons of direct extraction methods of microbial ADN from different paddy soils. Saudi Journal of Biological Sciences, 19, 337-342. [ Links ]
Robe, P., Nalin, R., Capellano, C., Vogel, T.M. & Simonet, P. (2003). Extraction of ADN from soil. European Journal of Soil Biology, 39,183-190. [ Links ]
Rives, N., Acebo, Y., & Hernández, A. (2007). EL CULTIVO DEL ARROZ Y SU IMPORTANCIA ECONÓMICA. Cultivos Tropicales. 28 (2), 29-38. [ Links ]
Wang, J., Ma, T., Zhao, L., Jinghua, L., Li, G., Liang, F., Liu, R. (2008). Comparison of methods for total community ADN extraction and purification from soilless substrate. World J Microbiol. 24,1981-1987. [ Links ]
Young, J., Rawlence, N & weyrich, L. (2014). Limitations and recommendatins for successful ADN extraction from foren-ce soil sample. Science and Justice, 54, 238-244. [ Links ]
Zaporozhenko, E.V., Slobodova, L.V., Bouly-gina, E. S., Kravchenko, K.I., & Kuznets-ov, B. B. (2006). Method for Rapid ADN Extraction from Bacterial Communities of Different Soils. Microbiology, 75(1), 127-134. [ Links ]
Tanase A., Mereuta L.Chiciudean L., Ionescu,R., Milea,L., Cornea C., Vassu T., Stoica L.,(2015). Comparison of Total DNA Extraction Methods for Microbial Community Form Polluted Soil, 6, 612- 622. [ Links ]