










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkGaceta Médica Boliviana
versión On-line ISSN 1012-2966
Gac Med Bol vol.40 no.2 Cochabamba dic. 2017
Caso Clínico
Evolución de un cuadro de hemorragia intracraneal en lactante con trombocitopenia inmune primaria
Evolution of a case of intracraneal hemorrhage in infant with primary immune trombocytopenia
Maiza Abujder Abu-khdeir1,d, Heydi Sanz Arrazola2,a, Ricardo Villegas Nava3,b, Enrique Gonzalo Rojas Salazar1,d.
1Facultad de Medicina, Universidad Mayor de San Simón, Cochabamba-Bolivia.
2Hospital del Niño Manuel Ascencio Villarroel, Cochabamba, Bolivia.
3Hospital Clínico Viedma, Cochabamba-Bolívia.
aMedica pediatra neuróloga; bMédico hematólogo; dMédico cirujano.*Correspodencia a:Maiza Abujder A. Correo electrónico: jsofiabujder@hotmail.com
Recibido el 25 de julio de 2017.
Aceptado el 8 octubre de 2017.
Resumen
La trombocitopenia inmune primaria (TIP) es una enfermedad hematológica de causa inmunológica que presenta una plaquetopenia inferior a 100 000 plaquetas/mm3 y tiene una incidencia de 4 casos por cada 100 000 habitantes, siendo más prevalente entre los 2 a 6 años de edad. Esta entidad continúa siendo un desafío con respecto a la terapéutica y puede conllevar a complicaciones graves muy difíciles de resolver una vez establecidas.Se presenta el caso de un lactante menor de sexo masculino que curso con por un cuadro de palidez generalizada con aparición súbita de hematoma en mucosa oral, máculas equimóticas y hemorragias puntiformes en toda la superficie corporal. En los exámenes realizados se evidenciaron trombocitopenia y anemia grave, llegando al diagnóstico de trombocitopenia inmune primaria inicialmente manejada con transfusiones y posteriormente con corticoides. A pesar del manejo, el cuadro evolucionó con complicaciones de hemorragia intraparenquimatosa y declino neurológico.
Palabras claves:trombocitopenia inmune primaria, hemorragia intraparenquimatosa, esplenectomía.
Abstract
Primary immune thrombocytopenia (PIT) is a hematological disease of immunological cause that has platelet count less than 100 000 platelets / mm3 and has an incidence of 4 cases per 100 000 inhabitants, being more prevalent between 2 and 6 years of age. This entity remains a challenge with regard to therapeutics and can lead to serious complications that are difficult to resolve once established. We report the case of a young male infant who has a generalized pallor with sudden onset of hematoma in the oral mucosa, equimotic macules and punctate hemorrhages throughout the body surface. In the examinations performed thrombocytopenia and severe anemia were evident, arriving at the diagnoses of idiopathic thrombocytopenic purpura initially managed with transfusions and later with corticoids. despite the treatment the case progress whith complications of intraparenchymal hemorrhage accompanied by neurological decline
Keywords:primary immune thrombocytopenia, intraparenchymal hemorrhage, splenectomy.
Se define a la trombocitopenia (TB) como una cifra de plaquetas inferior a 100 000 plaquetas/mm3 y se considera TB severa con un recuento menor a 50 000. Este trastorno hematológico afecta a pacientes de todas las edades, géneros y razas, pudiéndose presentar en forma aislada o asociada a una amplia gama de patologías1,2.
Con respecto a la estandarización actual de la terminología, las definiciones y los criterios de esta enfermedad, que fueron publicados el año 2009 en la revista “Blood”, actualmente la denominación de: “púrpura trombocitopénica idiopática” con su acrónimo PTI fue reemplazada por el nuevo término de: “trombocitopenia inmune primaria”3,4.
Esto se debe a que muchos pacientes no presentan púrpura durante la presentación clínica y además se sabe actualmente que la patogenia implica un mecanismo inmunológico (enfermedades autoinmunes y de autoagresión), por lo que no debería denominarse idiopática. Sin embargo el acrónimo “PTI” se mantiene vigente en algunas fuentes debido a su amplia difusión en la literatura médica1,2.
La literatura internacional reporta una incidencia de PTI de 4 casos por cada 100 000 habitantes, cuya mayor prevalencia oscila entre los 2 a 6 años5,6.
La causa de PTI es desconocida en la mayoría de los casos, pero puede ser provocada por vacunas, o la exposición a antígenos virales después de una infección respiratoria o gastrointestinal7-9.
La clasificación de esta enfermedad según su evolución es de dos tipos: transitoria o persistente. Suele ser transitoria, con una duración corta y una recuperación en menos de seis meses en el 70%, de los casos, generalmente en pacientes pediátricos; y en caso de ser persistente, en un 10% de los casos, la evolución es crónica con duración mayor a un año a partir de su presentación10-13.
Los metodos de diagnostico y las estrategias de tratamiento son imprecisos y pueden variar entre los diferentes pacientes. El diagnostico se basa en la historia clinica, examen fisico, contage global de celulas sanguineas y analisis del extendido de sangre periferica que debe contribuir para excluir otras causas de trombocitopenia. La medula osea en esta patologia es normal14-16.
La presentación clínica varía bastante acorde a cada paciente; existiendo niños asintomáticos o que con leves manifestaciones cutáneas hasta casos extremos que implican complicaciones hemorragias severas a nivel gastrointestinal o intracraneal, con un riesgo de 0,5 a 3% respectivamente, y raras veces existe compromiso de la vida del paciente.12,13 hemorragias opuntiformes (petequias) diseminadas por el organismo y sangramientos por las membranas mucosas. Las hemorragias pueden ser espontaneas o desencadenadas por traumas minimos16.
En la mayoría de los casos la resolución es espontánea y el tratamiento es de tipo profiláctico, sin embargo en casos graves con trombocitopenia severa y alto riesgo de complicaciones las opciones de tratamiento incluyen: inmunoglobulinas, anticuerpos anti-D, esplenectomia, e inmunosupresores que incluyen cortico esteroides como la metilprednisolona, que conllevan a la remisión de la enfermedad en muchos casos15,16.
De entre las complicaciones la hemorragia intracraneal se acompaña de peor pronostico. La mortalidad de los niños con PTI es de cerca del 1%, y en adultos puede alcanzar hasta el 5%17.
Presentación del caso
Se trata de un lactante menor de 1 mes y 21 días de edad de sexo masculino, eutrófico y con vacunas completas, que ingresa en fecha del 08-09-16; referido al servicio de emergencias del Hospital Materno Infantil German Urquidi (HMIGU), proveniente del Centro de Salud de Villa Israel, por un cuadro clínico de aproximadamente dos días de evolución caracterizado por presentar palidez generalizada con aparición súbita de máculas equimóticas y hemorragias puntiformes (petequias) en cara, cuadro que se exacerba en las siguientes 24 horas con aumento en el número y la extensión de las lesiones de forma generalizada en toda la superficie corporal, incluyendo: cabeza, tronco, abdomen y las cuatro extremidades (Figura 1).
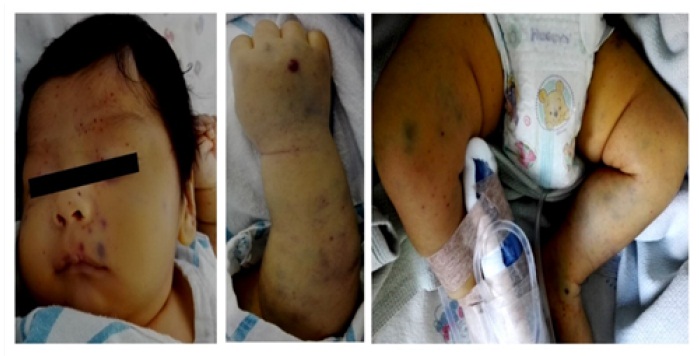
Figura 1:Examen físico del paciente, evidenciándose la presencia de petequias y zonas equimóticas en la región de la cara y las extremidades tanto superiores como inferiores
El lactante presenta signos vitales dentro de parámetros normales, sin alzas térmicas ni deposiciones líquidas. Posterior a la evaluación inicial se realiza la internación del paciente.
En la anamnesis no se evidenciaron antecedentes patológicos relevantes y como antecedentes perinatales no patológicos de interés se trató de un embarazo controlado parto por cesárea debido a DCP materna con obtención de neonato sano a término de 39 semanas de gestación con peso de 3,350 kg y talla de 51 cm, APGAR 8-9, PC: 35,5 cm, grupo sanguíneo tipo RH O+ tanto del paciente como de la madre, recibió lactancia materna y vacuna BCG, a la cual presento becegeítis.
Al examen físico se aprecia paciente en aparente regular estado general, activo reactivo a estímulos externos, con respiración espontánea y Glasgow de 15, buena tolerancia oral, piel y mucosas normo hidratadas con palidez generalizada a predominio de región facial y tórax anterior junto con equimosis y petequias de 1 a 3 mm, ojos con edema bipalpebral de leve intensidad, y a nivel de la boca se evidencia hematoma en el paladar duro.
Ante este cuadro clínico se decide administrar al paciente 500 ml de solución dextrosa al 5% por vía endovenosa, junto con 3 ml de cloruro de sodio y 2,5 ml de cloruro de potasio, junto con el control continuo de: signos vitales, curva térmica, estado de conciencia, signos de sangrado, diuresis horaria y balance hídrico. Posteriormente se realizan los siguientes exámenes complementarios: radiografía de tórax y abdomen, ecografía transfontanelar, extendido de sangre periférica, y laboratorios (incluyendo: grupo sanguíneo y factor Rh, hemograma completo, pruebas de coagulación, ionograma, proteinograma, glicemia, pruebas de función hepática y renal. LDH, PCR, TORCH, perfil de hierro, examen general de orina, hemocultivo, urocultivo, coprocultivo, coproparasitologico simple y moco fecal). El hemograma en fecha de ingreso reporta hemoglobina de 6,9 g/dl y plaquetopenia de 10 000 plaquetas/mm3 y en el examen de extendido periférico de sangre se tienen alteraciones de las tres series celulares, observándose: imágenes de rouleaux, hemolisis, linfocitos atípicos, y plaquetopenia (Figura 2).
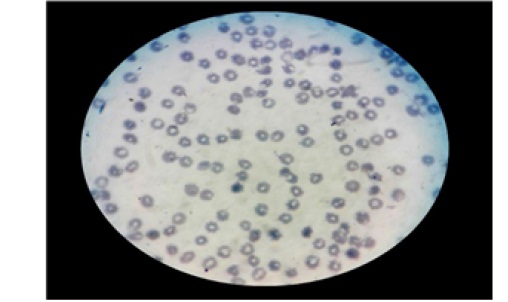
Figura 2:Extendido sanguíneo periférico con presencia de: hemolisis eritrocitaria, imágenes de rouleaux, linfocitos atípicos, y plaquetopenia con recuento plaquetario menor a 10 000/mm3
Con respecto a las pruebas de coagulación no presenta alteraciones, y resto de pruebas de laboratorio dentro de parámetros normales.
En base a los datos obtenidos de la anamnesis, la clínica y la confirmación por exámenes complementarios se llega a los diagnósticos de purpura trombocitopenica idiopática y anemia severa. Frente a este cuadro se realiza la valoración por hematología, indicándose la transfusión de 50 ml de concentrado de plaquetas (CPQ) a cada 8 horas, junto con 3 mg de vitamina K cada 24 horas, y también fueron transfundidos concentrados de glóbulos rojos.
Sin embargo debido a la falta de mejoría clínica y laboratorial al día siguiente se cambia el tratamiento, suspendiendo la transfusión de plaquetas, y se inicia la infusión de dexametasona endovenosa en forma lenta y diluida a dosis de 2,5 mg a cada 24 horas durante un periodo de tres días.
A pesar de esta nueva conducta a los cuatro días de tratamiento, desde el ingreso el lactante evoluciona desfavorablemente, sin lograr conciliar el sueño con normalidad, alternando periodos de irritabilidad con hiporreactividad y letargia, y aparición de picos febriles.
Frente a estas nuevas alteraciones el paciente es valorado por neurología, indicándose una ecografía transfontanelar, que reporta hemorragia intraparenquimatosa extensa a nivel de región parietotemporal izquierda sin desviación de línea media (Figura 3), y es indicando tratamiento profiláctico anticonvulsivante con fenobarbital endovenoso diluido a dosis de carga de 100 mg, seguido de dosis de mantenimiento de 12 mg a cada 12 horas.
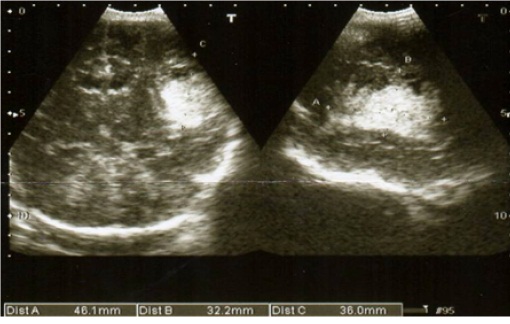
Figura 3:que evidencia hemorragia intraparenquimatosa extensa sin desviación de línea media, a nivel de la región parietotemporal izquierda, con las siguientes dimensiones: 4.6 de diámetro x 3.6 de altura x 3.2 cm de profundidad.
Frente a la falta de mejora del estado neurológico se realiza una tomografía axial computadorizada de cráneo, reportando hemorragia intraparenquimatosa parietotemporal izquierda con desvió de línea media MARSHAL III e hipertensión intracraneal (Figura 4).
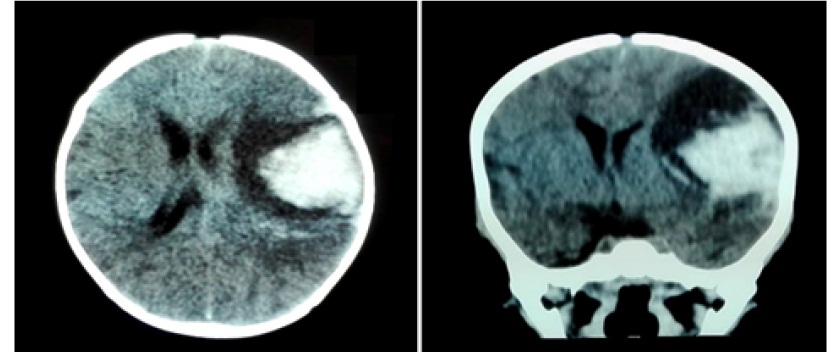
Figura 4:Tomografía simple de cráneo que evidencia hemorragia intraparenquimatosa a nivel de la región parietotemporal izquierda con desvió de la línea media MARSHAL III e hipertensión intracraneana.
Con estos resultados se indica intervención quirúrgica previa corrección de la plaquetopenia severa con transfusión pre, trans y postquirúrgica, y se procede a transfundir una unidad de plaquetas y 80 ml de concentrado de glóbulos rojos. Desde el ingreso del paciente se transfundieron en total: trece unidades de concentrados de plaquetas, tres unidades de glóbulos rojos y una unidad de plasma fresco congelado.
Sin embargo no se consigue mejorar la plaquetopenia y a los cinco días de internación el lactante presenta empeoramiento del estado neurológico con presencia de letargia, signos de hipertensión endocraneana, anisocoria pupilar, un estado de acidosis metabólica e inestabilidad hemodinámica con variabilidad en los signos vitales, llenado capilar alterado, saturación de oxigeno baja con necesidad de oxigeno suplementar, por lo que se procede al traslado a la unidad de cuidados intensivos pediátricos y la intubación oro traqueal.
En las horas siguientes se produce declino neurológico con puntuación de Glasgow de 5, pupilas midriáticas hiporreactivas denotando muy mal pronóstico y al día siguiente el paciente fallece a los cinco días de internación por muerte cerebral y anemia severa a causa de la hemorragia intraparenquimatosa y el síndrome de hipertensión endocraneal desencadenados a su vez por la progresión de una purpura trombocitopenia de origen desconocido.
Discusión
La PTI se caracterizada tanto por la destrucción acelerada de plaquetas como por la producción inadecuada de las mismas, dicha patología es mediada por auto anticuerpos dirigidos contra antígenos de la membrana plaquetaria, otro factor es la deficiente producción por el megacariocito debido a la acción de estos anticuerpos sobre la membrana del megacariocito en la médula ósea3,16.
La mayoría de los anticuerpos involucrados están dirigidos contra epítopes de glucoproteínas de la membrana plaquetaria, principalmente el complejo GPIIb/IIIa y GPIb/IX, estos se detectan en un 43-57% en los pacientes con PTI y la clase de inmunoglobulina involucrada es la IgG en el 92% de los casos3,17.
Las plaquetas opsonizadas por anticuerpos tipo IgG son destruidas por macrófagos en el bazo18 y uno de los beneficios del uso de dexametasona es que modula la respuesta inmunológica. El objetivo principal del tratamiento es revertir la trombocitopenia y de ese modo evitar complicaciones manteniendo la cifra de plaquetas por encima de 30 000/mm3 menores cifras implica riesgo de hemorragia grave, riesgo que es mayor en individuos de edad avanzada3.
El tratamiento más comúnmente usado para PTI es el denominado de primera línea y consiste en los glucocorticoides y la inmunoglobulina intravenosa, debemos tomar en cuenta que los esteroides deben ser suspendidos una vez que el conteo plaquetario se ha normalizado, o en quienes no han respondido en un lapso de cuatro semanas tras haber comenzado su consumo, ante dicha situación se recomienda retirar el fármaco y buscar otras alternativas de tratamiento19-21. En los pacientes que no responden adecuadamente al tratamiento de primera línea o en aquellos que presentan recaídas, o PTI crónica se recomienda el tratamiento de segunda línea que es la esplenectomía, dicho procedimiento no se recomienda antes de que transcurran seis meses desde el diagnóstico debido a la mejoría espontánea que se observa en la mayoría de los pacientes con PTI2.
En casos graves como ser el conteo plaquetario menor a 30.000/mm3 o ante la presencia de complicaciones (hemorragia gastrointestinal o intracraneal), se recomienda terapia inmediata que incluye la transfusión de plaquetas, inmunoglobulina intravenosa, y el uso glucocorticoides como la dexametasona20.
La esplenectomía puede llevarse a cabo con cifras bajas de plaquetas, aunque son deseables cifras por encima de 20000/mm3 para laparoscopia y laparotomía por encima de 80000/mm3, del mismo modo debe realizarse la vacunación del paciente frente a bacterias encapsuladas como Haemophilus influenza tipo B, Neisseria meningitidis y Streptococcus pneumoniae al menos dos semanas antes del procedimiento como medida profiláctica22.
En pacientes con contraindicación para la esplenectomía o con PTI crónica se llevaron a cabo estudios utilizando agonistas de los receptores de la trombopoyetina como ser la rominoplostim y eltrombopag dichos estudios publicados en la revista “The Lancet” obtuvieron buenos resultados para aumentar el recuento de plaquetas y reducir la hemorragia clínicamente significativa en los niños con PTI persistente o crónica23,24.
Actualmente se están realizando una serie de estudios con resultados prometedores mediante el uso del rituximab que es un anticuerpo monoclonal quimérico anti-CD20 obtenido por ingeniería genética, los estudios demostraron que éste se une específicamente a los linfocitos B, lo que conduce a la destrucción de los mismos22.
Con la eliminación de los linfocitos B cesa la síntesis de anticuerpos; lo que explica su efectividad en el tratamiento de algunas enfermedades auto inmunitario, y existen buenos resultados sobre su uso en la PTI en adultos. La experiencia sobre su empleo en la población pediátrica es muy escasa existiendo únicamente descripciones de casos aislados, mismos que dieron buena respuesta ante la PTI en pediatría25.
En relación a nuestro paciente, ante la sospecha de PTI se realizó la referencia a un centro de salud de tercer nivel al HMIGU, donde se brindó el tratamiento y los estudios necesarios acordes a la literatura, pese a todo se presentaron complicaciones tales como la hemorragia severa a nivel intracraneal, misma que está presente únicamente en el 1% de los niños con PTI, acorde a los casos descritos este tipo de hemorragias se asocian con elevadas tasas de morbilidad incluso con una adecuada y rápida intervención.
Cabe resaltar que los signos clínicos son muy importantes para tener un diagnóstico presuntivo de PTI por lo que deben ser tomados en cuenta al momento de evaluar a cualquier paciente y poder brindarle el tratamiento oportuno, puesto que mayoría de los casos la PTI al ser tratada presenta una evolución favorable.
Referencias bibliográficas
1. Astigarraga I. Actualización sobre trombocitopenias y trombocitopenia inmune primaria. Bol Soc Valencia Pediatr 2012; 44:19-24.
2. Rodeghiero F, Stasi R, Gernsheimer T, Michel M, Provan D, Arnold DM, et al. Standardization of terminology, definitions and outcome in immune thrombocytopenic purpura of adults and children: Report from an international working group. Blood 2009; 113:86-93.
3. García B, Espinosa L, Nava AH, Rubio B. Trombocitopenia inmune primaria. El Residente 2015; 10(3):154-65.
4. Stasi R, Amadori S, Osborn J, Newland AC, Provan D. Long-term outcome of otherwise healthy individuals with incidentally discovered borderline thrombocytopenia. PLoS Med 2006; 3(3):24.
5. Kistangari G, McCrae KR. Immune thrombocytopenia. Hematol Oncol Clin North Am 2013; 27 (3):495-520.
6. Lo E, Deane S. Diagnosis and classification of immunemediated thrombocytopenia. Autoimmun Rev 2014; 13(4):577-83.
7. Elena G, D’Aloi K, Veber S, Martinez G, Amaral D, Rosso D, y col. Trombocitopenia inmune en menores de cuatro meses: experiencia institucional. Hematologia 2015; 19(1):70-2.
8. Kühne T, Buchanan GR, Zimmerman S, Michaels LA, Kohan R, Berchtold W, Imbach P. A prospective comparative study of 2540 infants and children with newly diagnosed idiopathic thrombocytopenic purpura from the Intercontinental Childhood ITP Study Group. J Pediatr 2003; 143(5):605-8.
9. Imbach P, Kühne T, Müller D, Berchtold W, Zimmerman S, Elalfy M, Buchanan GR. Childhood ITP: 12 months follow-up data from the prospective registry I of the Intercontinental Childhood ITP Study Group (ICIS). Pediatr Blood Cancer 2006 Mar; 46(3):351-6.
10. Farhangi H, Ghasemi A, Banihashem A, Badiei Z, Jarahi L, Eslami G, et al. Clinical Features and Treatment Outcomes of Primary Immune Thrombocytopenic Purpura in Hospitalized Children under 2-Years Old. Iran J Ped Hematol Oncol 2015; l6 (1):24-33.
11. Lo C, Wong W, Glader B, Jeng M. Immune Thrombocytopenia in Children Less Than 1 Year of Age: A Singleinstitution 10-year Experience. Pediatr Hematol Oncol 2013; 35(5):406-8.
12. Donato H y col. Púrpura trombocitopenia inmunitaria. Guía de diagnóstico y tratamiento Arch Argent Pediatr 2010; 108(2):173-8
13. Sandoval C, Visintainer P, Ozkaynak MF, Tugal O, Jayabose S. Clinical features and treatment outcomes of 79 infants with immune thrombocytopenic purpura. Ped Blood & Cancer 2004; 42(1):109-12.
14. Rodeghiero F, Stasi R, Gernsheimer T. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood 2009; 113(11):2386-93.
15. Dominguez MV, Rodríguez H. Cellular and biochemical mechanisms involved in physiopathogenesis of autoimmune thrombocytopenic purpura. Gac Med Mex 2002; 138(5):461-72.
16. Carson KR, Evens AM, Richey EA, Habermann TM, Focosi D, Seymour JF, et al. Progressive multifocal leukoencephalopathy after rituximab therapy in HIV-negative patients: a report of 57 cases from the Research on Adverse Drug Events and Reports project. Blood 2009; 113(20): 4834-40.
17. Kashiwagi H, Tomiyama Y. Pathophysiology and management of primary immune thrombocytopenia. Int J Hematol 2013; 98(1):24-33.
18. Zaja F, Battista ML, Pirrotta MT, Palmieri S, Montagna M, Vianelli N, et al. Lower dose rituximab is active in adult patients with idiopathic thrombocytopenic purpura. Hematological 2008; 93(6):930-3.
19. Neylon AJ, Saunders PW, Howard MR, Proctor SJ, Taylor PR. Clinically significant newly presenting autoimmune thrombocytopenic purpura in adults: a prospective study of a population-based cohort of 245 patients. Br J Haematol 2003; 122(6):966-74.
20. Neunert C, Lim W, Crowther M, Cohen A, Solberg L, Crowther MA. Evidence-based practice guideline for immune thrombocytopenia. Blood 2011; 117(16):4190-207.
21. Sanz MA, Vicente V, Fernández A, López MF, Grande C, Jarque I, y col. Diagnóstico, tratamiento y seguimiento de la trombocitopenia inmune primaria. Med Clínica 2012; 138(6):261.
22. James B, Bussel MD. Eltrombopag for the treatment of children with persistent and chronic immune thrombocytopenia (PETIT): a randomised, multicentre, placebo-controlled study. The Lancet 2015; 8(2):315-25.
23. Michael D, Tarantino MD. Romiplostim in children with immune thrombocytopenia: a phase 3, randomised, double-blind, placebo-controlled study. The Lancet 2016;388(1):45-54.
24. Cindy E. Neunert. Current management of immune thrombocytopenia. American Society of Hematology 2013; (15):276-82.
25. Liang Y, Zhang L, Gao J, Hu D, Ai Y. Rituximab for children with immune thrombocytopenia: A systematic review. PLoS One 2012; 7(5):366-98.

